Invited Symposium: SERCA-Type of Calcium Pumps and Phospholamban
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Results
Topology of calcium binding
Original experiments on site directed mutagenesis of transmembrane helices suggested that six residues (Glu309, Glu771, Asn796, Thr799, Asp 800 and Glu908, as shown in Fig 1D) may be involved in Ca2+ binding (Clarke et al., 1989). This suggestion was based on the interference of single mutations on the normally observed Ca2+ inhibition of reverse enzyme phosphorylation by Pi. Direct measurements of Ca2+ binding (or lack thereof) by these mutants was prevented by the limited quantities of recombinant protein obtained by transfection of cell cultures. In fact, the prevailing methods (e.g., calcium phosphate, DEAE-Dextran or liposomes) for transfection with plasmids containing wild type or mutated cDNA, affect only a very small percentage of cells in culture (Fig 2). Therefore the overall expression of heterologous protein is quite limited.
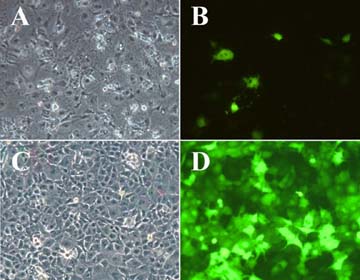 Fig.
2 - Phase (A and C) and fluorescence (B and D) micrographs of COS-1 cells
expressing Enhanced Green Fluorescence Protein (EGFP) Comparison of
EGFP gene transfer efficiency following transfection by DEAE-Dextran and
Chloroquine shock, or following infection with recombinant adenovirus vector
(C and D), under optimal conditions for each method. Note the much larger
percentage of cells expressing the hexogenous gene in the infected sample.
Fig.
2 - Phase (A and C) and fluorescence (B and D) micrographs of COS-1 cells
expressing Enhanced Green Fluorescence Protein (EGFP) Comparison of
EGFP gene transfer efficiency following transfection by DEAE-Dextran and
Chloroquine shock, or following infection with recombinant adenovirus vector
(C and D), under optimal conditions for each method. Note the much larger
percentage of cells expressing the hexogenous gene in the infected sample.
We found that transfer of heterologous cDNA can be made much more efficient by the use of recombinant adenovirus vectors, whereby the entire population of COS-1 cells in culture is infected and much larger quantities of recombinant protein are produced (Fig 2). The infected cells are then harvested and homogenized for preparation of microsomal fractions. These microsomes contain recombinant SERCA protein at a concentration approximately one tenth as high as that of the native ATPase in sarcoplasmic reticulum vesicles obtained from rabbit skeletal muscle. This concentration is sufficient for measurements of Ca2+ binding by isotopic tracer and filtration methods, in the presence of low (10-20 micromolar) Ca2+. The Ca2+ binding levels per mg of microsomal protein, related to the stoichiometry of expressed ATPase by a number of immunological and functional titrations, are consistent with binding of two Ca2+ per ATPase, as previously established with native sarcoplasmic reticulum ATPase.
The effects of the original Clarke et al. (1989) mutations on Ca2+ binding are shown below in Fig 3. It is of interest that while most mutations interfere with binding of both calcium ions (at least in the presence of low Ca+), the Glu309Gln and the Asn796Ala mutations reduce Ca2+ binding by approximately 50%, raising the possibility that inhibition by these two mutations affects only one of the two calcium ions that are normally bound.
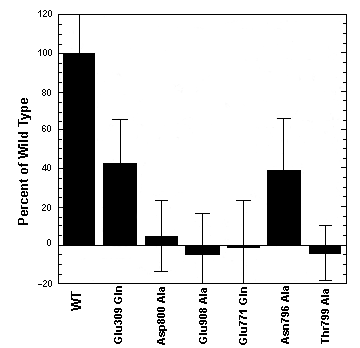 Fig.
3 - Effects of various mutations on Ca2+ binding by recombinant ATPase
Binding was measured at pH 7.0, in the presence of 20 uM Ca2+. 100% correspond
to 2 Ca2+/ATPase (Strock et al., 1998).
Fig.
3 - Effects of various mutations on Ca2+ binding by recombinant ATPase
Binding was measured at pH 7.0, in the presence of 20 uM Ca2+. 100% correspond
to 2 Ca2+/ATPase (Strock et al., 1998).
Parallel experiments on enzyme phosphorylation with Pi (at pH 6.2) reveal that while Glu309Gln (and other mutations) interfere completely with Ca2+ inhibition of the Pi reaction, the Asn796Ala mutation does not. This differential behavior suggests that Glu309Gln and Asn796Ala inhibit binding of an alternative Ca2+, and that binding of only one Ca2+ (affected by the Glu309Gln mutation) is required for inhibition of the Pi reaction. Even more interesting is the fact that at pH 7.0 (rather than pH 6.2) even the Glu309Gln mutation does not interfere with Ca2+ inhibition of the Pi reaction (Fig. 4). This suggests that ionization of acidic functions of other participating residues (i.e., Glu771 and/or Asp800) occurs as the pH is shifted from 6.2 to 7.0, thereby facilitating binding of inhibitory Ca2+ by the Glu309Gln mutant. On the other hand, in the Asn796Ala mutant, the presence of the Glu309 acidic function allows binding of inhibitory Ca2+ even at pH 6.2. This is consistent with specific roles of the various residues participating in complexation of the two calcium ions. It is also consistent with the role of acidic residues in the observed exchange of 2 Ca2+ with 2 H+ during the ATPase cycle (Chiesi and Inesi, 1980).
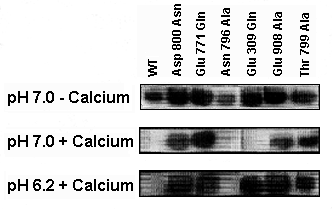 Fig.
4 - ATPase phosphorylation by Pi The level of phosphoenzyme was detected
by autoradiography. Note that all mutants show phosphorylation in the absence
of Ca2+. The phosphorylation reaction is inhibited by Ca2+ (20 uM) when
WT enzyme and the Asn796Ala mutant are used at pH 7.0 and 6.2, and when
the Glu309Gln mutant is used at pH 7.0. Ca2+ does not inhibit in all other
cases (Strock et al., 1998).
Fig.
4 - ATPase phosphorylation by Pi The level of phosphoenzyme was detected
by autoradiography. Note that all mutants show phosphorylation in the absence
of Ca2+. The phosphorylation reaction is inhibited by Ca2+ (20 uM) when
WT enzyme and the Asn796Ala mutant are used at pH 7.0 and 6.2, and when
the Glu309Gln mutant is used at pH 7.0. Ca2+ does not inhibit in all other
cases (Strock et al., 1998).
It should be pointed out that extensive experimentation on the Pi reaction have demonstrated that it is ultimately possible to inhibit phosphorylation of all these mutants with Pi if the Ca2+ concentration is raised to the mM levels. This indicates that no single mutation is totally disruptive of binding, but it is more or less effective in lowering the affinity of the site for one or both Ca2+. This is consistent with coordination of Ca2+ by several oxygens whose electronegativity may be either shared by the two Ca2+, or directed prevalently to one of the two Ca2+.
Topology of the thapsigargin binding site
The chemical structure of thapsigargin is well characterized (review: Christensen et al. 1997), and its inhibitory effect on intracellular Ca2+ ATPases was first observed by Thastrup et al. (1989). Its stoichiometric interaction with the sarcoplasmic reticulum Ca2+ ATPase, and its inhibitory effect at subnanomolar concentrations were then demonstrated (Sagara and Inesi, 1991; Lytton et al., 1991). Thapsigargin forms a dead-end complex with the ATPase, stabilizing a conformational state that favors bidimensional ordering of the ATPase protein within the plane of the membrane (Sagara et al.(1992a; 1992b; Witcome et al., 1992). Experimentation with fluorescent and azido-derivatives of thapsigargin suggests that the inhibitor adheres to the membrane, and interacts with neighboring ATPase domains (Hua et al., 1995; Hua and Inesi, 1997).
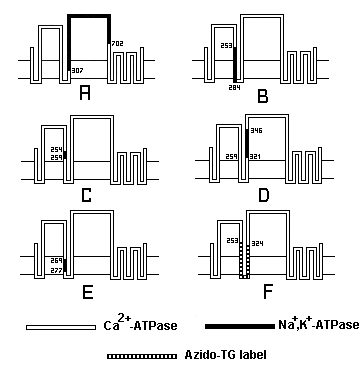 Fig.
5 - Chimeric exchanges between Ca2+ ATPase and Na+,K+ ATPase A: Exchange
of the large cytosolic region. B: S3,M3 exchange. C: S3 exchange. D:S4
exchange. E: M3 exchange. F: peptide segment (dark) covalently labeled
with an azido derivative of TG. Note that the S3 exchange (C), by itself,
reduced by three orders of magnitude the affinity of the ATPase for thapsigargin
(Zhong and Inesi, 1998).
Fig.
5 - Chimeric exchanges between Ca2+ ATPase and Na+,K+ ATPase A: Exchange
of the large cytosolic region. B: S3,M3 exchange. C: S3 exchange. D:S4
exchange. E: M3 exchange. F: peptide segment (dark) covalently labeled
with an azido derivative of TG. Note that the S3 exchange (C), by itself,
reduced by three orders of magnitude the affinity of the ATPase for thapsigargin
(Zhong and Inesi, 1998).
The specificity of thapsigargin is such that while total inhibition of the SERCA enzyme isoforms is produced, no inhibition of the Na+,K+ ATPase is observed. Taking advantage of such a specificity, chimeric constructs of the SERCA and Na+,K+ ATPases can be produced to test the involvement of various enzyme domains in the sensitivity of the SERCA ATPases to thapsigargin. It is found that exchange of the large and small cytosolic loops of the Ca2+ ATPase with the corresponding loops of the Na+,K+ ATPase yields an enzyme that retains Ca2+ dependence and thapsigargin sensitivity. These experiments prove that the cytosolic (i.e., extramembranous) loops do not participate in Ca2+ and/or thapsigargin binding (Sumbilla et al., 1993). However, exchanges in the M3-S3 (Norregaard et al., 1993) reveal that the S3 stalk segment is specifically involved (Zhong and Inesi, 1998) in conferring thapsigargin sensitivity to the SERCA ATPases (Figs. 5 and 6).
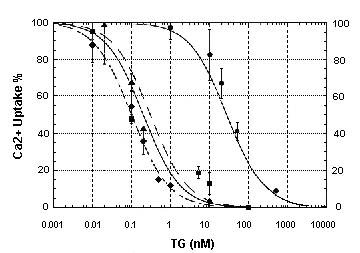 Fig.
6 - Concentration dependence of thapsigargin inhibition of Ca2+ transport
by WT ATPase and various chimeric constructs The velocities of ATP
dependent Ca2+ transport were measured, and the velocities observed in
the presence of thapsigargin were expressed as percentages of the velocity
sustained by each enzyme in the absence of thapsigargin. (triangle): WT
ATPase; (circle): S3 chimera (C in Fig 6); (square): M3 chimera (E in Fig
6); (diamond): S4 chimera (D in Fig 6), (Zhong and Inesi, 1998).
Fig.
6 - Concentration dependence of thapsigargin inhibition of Ca2+ transport
by WT ATPase and various chimeric constructs The velocities of ATP
dependent Ca2+ transport were measured, and the velocities observed in
the presence of thapsigargin were expressed as percentages of the velocity
sustained by each enzyme in the absence of thapsigargin. (triangle): WT
ATPase; (circle): S3 chimera (C in Fig 6); (square): M3 chimera (E in Fig
6); (diamond): S4 chimera (D in Fig 6), (Zhong and Inesi, 1998).
Since there is no homology (except for one single amino acid) in the S3 segments of the Ca2+ ATPase and the Na+,K+ ATPase, the effects of several single and/or stepwise mutations within the S3 segment can be evaluated. Such studies (Zhong and Inesi, 1998) reveal a strong reduction of the Ca2+ ATPase affinity for thapsigargin when Gly257 is mutated to the corresponding Na+,K+ ATPase residue (Ile). There is a greater reduction when Gln259 is mutated to Leu in addition to the 257 mutation, although single mutation of Gln259 (by itself) is much less effective. It is of great interest that mutation of Phe256 (the only homologous residue in the Ca2+ ATPase and the Na+,K+ ATPase in the S3 segment) to eliminate its ring moiety, also reduces the affinity of the Ca2+ ATPase to thapsigargin (Yu et al., 1998). Since the Na+,K+ ATPase is not sensitive to thapsigargin, the mutational analysis suggests that Phe256 interacts with thapsigargin only if permitted specifically by Gly257 and Gln259.
Extensive studies on chemical modification of the thapsigargin structure (review: Christensen et. al., 1997) have demonstrated that the most critical regions of thapsigargin for inhibition of Ca2+ ATPase are the side chains on C3 and C8. Inversion of the side chain at C8 decreases the affinity of thapsigargin for the ATPase by three orders of magnitude. Inversion of the side chain on C3 decreases the affinity by two orders of magnitude. In Figure 7 the critical side chains of the thapsigargin structure, and of the critical amino acids (Phe256, Gly257 and Gln259) of the ATPase S3 segment (slightly above the cytosolic surface of the membrane) are highlighted in green. The molecular graphics representation of these structures, raises the possibility that partitioning of the long hydrophobic chain provides the means of thapsigargin adherence to the membrane, while interaction of the critical side chains (green) with Phe256 (permitted by the neighboring Gly and Gln) provides specific stabilization.
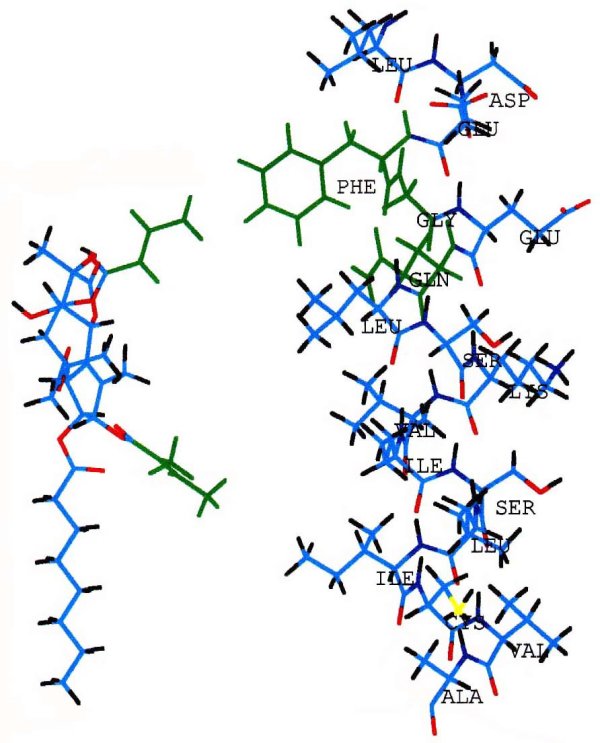
Fig. 7 - Molecular graphics matching thapsigargin with the S3 stalk segment of the ATPase Thapsigargin (left) with its side chains at C3 (lower) and C8 (upper) highlighted in green. These hydrophobic side chains extends downward in this view. The helix shown on the right side of the figure represents the S3 stalk segment of the ATPase, including Phe256 and Gly257 (highlighted in green), and the initial part of the M3 intramembrane region. The cytosolic membrane surface may be at the Ser(261)-Lys(262) level.
| <= Materials & Methods | RESULTS | Discussion & Conclussions => |
| Discussion Board | Next Page | Your Symposium |