Invited Symposium: Stroke/Cerebral Vasospasm
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Several types of cerebrovascular pathology can impair cerebral blood flow and cause a stroke. The most common form of stroke occurs as a result of the intraluminal occlusion of a cerebral vessel, which creates a focal zone of cerebral ischemia. Some occluded vessels will recanalize spontaneously after a period of occlusion, leading to reperfusion of an ischemic zone. The advent and approval of 'clot-busting' drugs for treating occlusive stroke increases the probability of vascular reperfusion in patients with occlusive stroke. Reperfusion following ischemia presents several challenges that can aggravate injury in compromised tissue (e.g. Bazan et al., 1992). It is therefore of increasing importance to identify mechanisms of reperfusion injury and to develop effective strategies to limit this type of injury.
The cerebral microvasculature represents an important target for treating reperfusion injury. Cerebral blood flow exhibits a biphasic response after reperfusion, characterized by an initial brief period of hyperemia followed by a delayed and prolonged period of hypoperfusion (Hossmann et al., 1973; Nemoto et al., 1975; Miller et al., 1980; Kagstrom et al., 1983; Shigeno et al., 1985). Delayed hypoperfusion can result in a secondary ischemic challenge to compromised tissue that aggravates ischemic injury. The cerebral microvasculature located downstream from a reperfused vessel plays a key role in delayed hypoperfusion (Hossmann et al., 1973; Schmidt-Kastner et al., 1987). Downstream microvessels become ischemic and often dilate during ischemia.
This ischemia-induced vasodilation probably contributes to the initial hyperemic response observed when reflow is established. The vessels subsequently constrict beyond baseline levels creating a secondary ischemic event that can persist for hours. Delayed, postischemic hypoperfusion thus produces a prolonged secondary period of ischemia that can aggravate pre-existing damage and/or elicit additional ischemic injury. Because of the potential importance of delayed vasoconstriction in the elaboration of secondary cerebral injury, a series of studies was undertaken to examine the mechanism(s) underlying delayed microvascular constriction following transient metabolic insufficiency. The present study examined the possible role of calcium-activated proteolysis, by the protease calpain in delayed vasoconstriction after transient hypoxia.
Materials and Methods
Cerebral microvessels were examined by utilizing a simplified experimental system in which live parenchymal microvessels can be visualized in in vitro brain slices (Sagher et al., 1993). Computerized videomicrosopy was used to visualize microvessels, and the intraluminal diameter of the vessels was measured at one-minute intervals during the course of each experiment. Studies using this technique have shown that microvessels in brain slices exhibit typical responses to a variety of vasodilators and vasoconstrictors (Sagher et al., 1993; Jin et al., 1994; Fergus et al., 1995, 1996, 1997a,b). The vessels are also capable of repeated constriction and dilation when treated with appropriate vasoactive agents. Parenchymal microvessels in the brain slice preparation thus retain functional responses despite the absence of intraluminal flow.
In the present study, slices were subjected to one hour of moderate hypoxic conditions resembling those observed in an ischemic penumbra (Chen et al., 1996). To test the possible contribution of calpain to hypoxia-induced alterations in cerebral microvessels, hypoxia was administered in the presence or absence of the calpain inhibitor MDL28170 (100 micromolar). After one hour of hypoxic conditions, the slices were re-oxygenated and monitored until a stable diameter is recorded; this typically required an hour or two after re-oxygenation.
Results
An example of a cerebral microvessel monitored in hippocampal brain slice is shown in Figure 1. The intraluminal borders of the vessel are visible as it runs diagonally across the image; a pyramidal cell body is also observed (arrow).
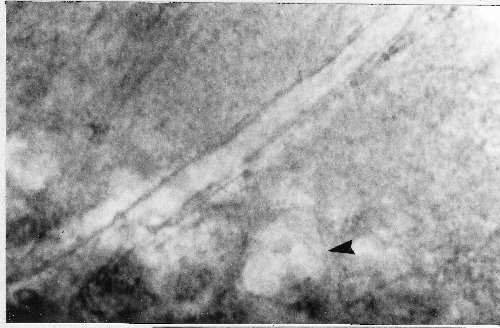 Fig1: A microvessel in a hippocampal slice. The microvessel runs diagonally across the image, and is located in the strata pyramidale and radiatum of region CA1. The intraluminal diameter of the vessel is 12 micrometers, and the cell body of a neighboring CA1 pyramidal cell can also be seen (arrow; see Sagher et al., 1993 for details).
Fig1: A microvessel in a hippocampal slice. The microvessel runs diagonally across the image, and is located in the strata pyramidale and radiatum of region CA1. The intraluminal diameter of the vessel is 12 micrometers, and the cell body of a neighboring CA1 pyramidal cell can also be seen (arrow; see Sagher et al., 1993 for details).
Transient hypoxia induced a biphasic response in cerebral microvessels (Figure 2). The vessels dilate in response to hypoxia, and remain dilated during the hypoxic period. Approximately 20 minutes after reoxygenation, the vessels constrict to levels beyond baseline and typically maintain this constriction for at least several hours. The hypoxia-induced alterations in cerebral microvessels thus mimic the types of vascular changes observed in response to ischemia in vivo.
Specifically, a biphasic response is observed that is characterized by an initial vasodilation followed by a delayed and prolonged vasoconstriction. These findings indicate that the brain slice technique can be used as a simple experimental system for examining the cellular and molecular mechanisms of the biphasic microvascular response to transient metabolic insufficiency. In the presence of a calpain inhibitor (MDL28170) delayed posthypoxic constriction was abolished while the initial hypoxic vasodilation was not significantly altered. The average diameter of control posthypoxic vessels was approximately 70% of baseline, which represents a substantial posthypoxic constrictor response. In contrast, the average posthypoxic diameter of vessels treated with MDL28170 was approximately 110% of baseline, representing a complete blockade of delayed, posthypoxic vasoconstriction.
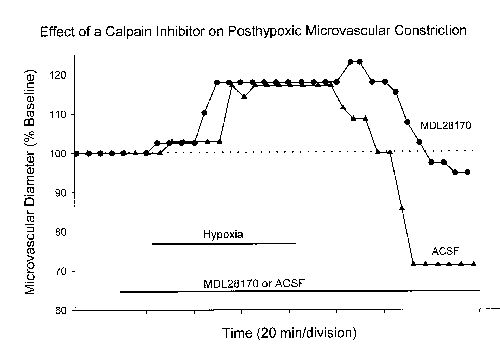 Fig2: The effect of the calpain inhibitor MDL28170 (100 mM) on posthypoxic vasoconstriction in cerebral microvessels. Two experiments are shown in which microvessels from hippocampal slices were subjected to moderate hypoxia for 60 minutes. The graphs show the time course of changes in microvascular diameter expressed as a percentage of the pre-hypoxia baseline; both vessels were preconstricted with an inhibitor of nitric oxide synthase (L-NAME) prior to establishing baseline measurements. The calpain inhibitor blocked posthypoxic vasoconstriction while the initial vasodilation was relatively unaffected. Abbreviation: ACSF=artificial cerebrospinal fluid.
Fig2: The effect of the calpain inhibitor MDL28170 (100 mM) on posthypoxic vasoconstriction in cerebral microvessels. Two experiments are shown in which microvessels from hippocampal slices were subjected to moderate hypoxia for 60 minutes. The graphs show the time course of changes in microvascular diameter expressed as a percentage of the pre-hypoxia baseline; both vessels were preconstricted with an inhibitor of nitric oxide synthase (L-NAME) prior to establishing baseline measurements. The calpain inhibitor blocked posthypoxic vasoconstriction while the initial vasodilation was relatively unaffected. Abbreviation: ACSF=artificial cerebrospinal fluid.
Discussion and Conclusion
The current results indicate that calpain-mediated proteolysis contributes to delayed vasoconstriction following metabolic insufficiency. In addition, the findings suggest that calpain inhibitors may be capable of attenuating secondary ischemia associated with reperfusion injury. In occlusive stroke, a cerebral vessel becomes blocked intraluminally providing a direct ischemic insult to downstream structures. The early re-establishment of blood flow in occluded vessels is essential for salvaging ischemic tissue in these patients; however, reperfusion triggers another complex set of events (e.g. delayed hypoperfusion) that can result in a secondary phase of ischemia and brain injury. Calpain inhibitors may prove to be of particular value in limiting secondary injury via their actions on the cerebrovasculature. Previous findings show that calpain inhibitors can inhibit the development and maintenance of cerebral vasospasm in animal models of subarachnoid hemorrhage (Minami et al., 1992; Cappelletto et. al., 1997).
Calpain inhibitors also attenuate ischemic brain injury in animal models of occlusive stroke (Hong et al., 1994; Bartus et al., 1994), and it is plausible that this protective effect is due in part to the amelioration of delayed cerebrovascular hypoperfusion. Although more speculative in nature, an effect of calpain inhibitors on vascular targets could also help explain their protective influence against secondary injury following traumatic damage to the brain and spinal cord (Saatman et al., 1996; Banik et al., 1997; Posmantur et al., 1997). Clearly, the role of calpain on the cerebral microvasculature in the context of ischemic or traumatic injury will need to be assessed directly in intact preclinical models of CNS injury. Nonetheless, the current results raise the possibility that the neuroprotective effects of calpain inhibitors in the in vivo setting are due, in part, to actions on vascular, as well as neuronal, elements.
References
- Banik, N.L., Matzelle, D., Gantt-Wilford, G. and Hogan, E.L. 1997. Role of calpain and its inhibitors in tissue degeneration and neuroprotection in spinal cord injury. Ann. N.Y. Acad. Sci. vol. 825, Neuroprotective Agents: Third International Conference ed. W. Slikker Jr. and B. Trembly, pp. 120-127. New York, The New York Academy of Sciences.
- Bartus, R., Baker, K., Heiser, A., Sawyer, S., Dean, R., Elliott, P. and Straub, J. 1994. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J. Cereb. Blood Flow Metab. 14: 537-544.
- Bazan, N.G., Brauquet, P. and Ginsberg, M.D. 1992. Neurochemical Correlates of Cerebral Ischemia. New York: Plenum.
- Cappelletto, B., Caner, H.H., Schottler, F., Kwan, A-L., Eveleth, D., Foley, P.L., Kassell, N.F., Lee, K.S. Attenuation of vasospasm and hemoglobin-induced constriction in the rabbit basilar artery by a novel protease inhibitor. Neurosurgical Focus 3: 1-13.
- Chen, Z.-F., Schottler, F., Arlinghaus, L., Kassell, N.F. and Lee, K.S. 1996. Hypoxic neuronal damage in the absence of hypoxic depolarization in hippocampal slices: the role of glutamate receptors. Brain Res. 708: 82-92.
- Fergus, A., Jin, Y., Thai, Q.-A., Kassell, N.F. and Lee, K.S. 1995. Vasodilatory actions of calcitonin gene-related peptide and nitric oxide in parenchymal microvessels of the rat hippocampus. Brain Res. 694: 78-84.
- Fergus, A., Jin, Y., Thai, Q.-A., Kassell, N.F. and Lee, K.S. 1996. Tonic protein kinase c-mediated vasoconstriction is unmasked when nitric oxide synthase is inhibited in cerebral microvessels. Neuroscience 74: 927-934.
- Fergus, A. and Lee, K.S. 1997a. Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res. 754: 34-45.
- Fergus, A. and Lee, K.S. 1997b. GABAergic regulation of cerebral microvascular tone in the rat. J. Cereb. Blood Flow Metab. 17: 992-1003.
- Hong, S., Goto, Y., Lanzino. G., Soleau, S. Kassell, N., and Lee, K., 1994. Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia, Stroke 25: 663-669.
- Hossmann, K-A., Lechtape-Gruter, H. and Hossmann, V. 1973. The role of cerebral blood flow for the recovery of the brain after prolonged ischemia. Z. Neurol. 204: 281-299.
- Jin, Y., Sagher, O., Thai, Q.-A., Kassell, N.F. and Lee, K.S. 1994. The effects of papaverine on phorbol dibutyrate-induced vasoconstriction in brain slice microvessels. J. Neurosurg. 81: 574-578.
- Kagstrom, E., Smith, M.L. and Siesjo, B.K. 1983. Local cerebral blood flow in the recovery period following complete cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 3: 170-182.
- Mehdi, S. 1991. Cell-penetrating inhibitors of calpain, TIBS 164: 150-153.
- Miller, C.L., Lampard, D.G., Alexander, K., and Brown, W.A. 1980. Local cerebral blood flow following transient cerebral ischemia. I. Onset of impaired reperfusion within the first hour following global ischemia. Stroke 11: 534-541.
- Minami, N., Tani, E., Maeda, Y., Yamaura, I. and Fukami, M. 1992. Effects of inhibitors of protein kinase C and calpain in experimental delayed cerebral vasospasm. J. Neurosurg. 76: 111-118.
- Nemoto, E.M., Snyder, J.V., Carroll, R.G. and Morita, H. 1975. Global ischemia in dogs: cerebrovascular CO2 reactivity and autoregulation. Stroke 6: 425-431.
- Posmantur, R., Kampfl, A., Siman, R., Liu, J., Zhao, X., Clifton, G.L and Hayes. R.L. 1997. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neurosci. 77: 875-888.
- Saatman, K.E., Murai, H., Bartus, R.T., Smith, D.H., Hayward, N.J., Perri, B.R. and McIntosh, T.K. 1996. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc. Natl. Acad. Sci. (USA) 93: 3428-3433.
- Sagher, O., Zhang, X.Q., Szeto, W., Thai, Q.-A., Jin, Y., Kassell, N.F. and Lee, K.S. 1993. Live computerized videomicroscopy of cerebral microvessels in brain slices. J. Cereb. Blood Flow Metab. 13: 676-682.
- Schmidt-Kastner, R., Hossmann, K-A. and Grosse-Ophoff, B. 1987. Pial artery pressure after one hour of global ischemia. J. Cereb. Blood Flow Metab. 7: 109-117.
- Shigeno, T., Teasdale, G.M., McCulloch, J., and Graham, D.I. 1985. Recirculation model following MCA occlusion in rats. Cerebral blood flow, cerebrovascular permeability and brain edema. J. Neurosurg. 63: 272-277.
| Discussion Board | Previous Page | Your Symposium |