Invited Symposium: Stroke/Cerebral Vasospasm
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Vascular smooth muscle (VSM) has a key role in the maintenance of haemodynamic and metabolic tissue homeostasis, responding to central and local signals. Whilst central control, mediated by neural or humoral pathways, can modulate local metabolite signals for periods of time, in physiological conditions local control mechanisms will usually prevail, and tissue homeostasis is maintained. In pathological conditions, such as vasospasm, failure of normal homeostatic mechanisms occurs resulting in tissue ischa emia, infarction and cell death.
Many pathological mechanisms have been proposed for the pathogenesis of cerebral vasospasm (MacDonald et al., 1991; Weir, 1995). Metabolic disorders such as excessive secretion of adrenergic compounds, to accumulation o f intracellular calcium (Kim et al., 1996) have also been suggested as causes of vasospasm. The duration of their presence is usually short term and the time course does not correlate with lumenal narrowing (3-10 days; Weir et al., 1978). Whilst many of t hese proposed mechanisms have been shown to be relevant to certain smaller mammals (Symon, 1978), application of these ideas to clinical trials have been inconclusive. Treatment with oral Nimodopine (Nomotop tm, Bayer) in one large prospective randomised trial (Pickard et al., 1989) demonstrated improvement in neurological outcome but no change in the lumen of the cerebral arteries on angiography (Petrok, et al., 1988). Suggesting therefore, that the clinical improvement was not due to this voltage depend ent calcium channel blocker preventing or reversing the vasospasm, but by improving outcome via some other means.
High intracellular ADP levels have been shown to alter the ability of VSM to relax (Fuglsang et al., 1993; Nishiye, 1993; Clark et al, 1994 ). ADP is normally maintained at low intracellular levels by stimulating mitochondria and increasing oxidative phosphorylation. The vessel’s energy metabolism, therefore, plays an important role in maintaining contractility by controlling [ADP] (Clark, 19 94). Alterations in intracellular energy metabolism may contribute to cerebral vasospasm by permitting elevated levels of ADP. For intracellular [ADP] to rise sufficiently to block relaxation either; 1) the mitochondria must be failing to respond to the intracellular [ADP] due to mitochondrial dysfunction, 2) oxidative phosphorylation must be uncoupled, or 3) ATPase activity (resulting in ADP production) must exceed mitochondrial oxidative capacity. In this study we have tested the hypothesis that energy metabolism has a role in cerebral vasospasm.
Materials and Methods
Tissue Preparation
The experiments used porcine carotid artery from adult pigs, harvested from a nearby abattoir within 15 minutes of death. The tissue was cooled to 4°C in physiological saline solution (PSS), stripped of loose adventitia, connective tissue and endotheli um and stored at 4°C overnight. A 6 ml well with stirrer and Clark oxygen electrode (Yellow Springs, Ohio USA) connected to a chart recorder was used to measure oxygen consumption (Clark et al., 1993). Several changes in buffer were used to maintain obser vations of up to 5 hours at 30°C. After each experiment the carotid was dried and weighed and oxygen consumption rates calculated assuming the solubility of oxygen to be 0.21 uMol/ml, after equilibrating the buffer with ambient oxygen (Clark et al., 1993) . Human CSF was collected with local ethics approval. Cerebrospinal fluid from subarachnoid haemorrhage patients was collected either via a ventricular drain placed for therapeutic reasons, or at the time of surgery. The patient was considered to have vas ospasm at the time of CSF sampling if arterial narrowing was seen on the diagnostic angiograme or a delayed neurological deficit developed during the period of hospitalisation, and CT scanning had excluded other causes, such as infarct, haematoma and hyd rocephalus. In the absence of arterial narrowing on the diagnostic angiograme, and absence of delayed neurological deficits, the patient was considered to non-vasospastic.
Oxygen consumption experiments
Filtered physiological saline was added to the stirred chamber, the Clark oxygen electrode introduced and control recordings taken. Strips of porcine carotid artery were placed in the chamber and allowed to equilibrate for 30 minutes to verify tissue v iability and to establish a baseline. A 200 mL aliquot of SAH CSF was then added to the 6 ml chamber (1:30 dilution of CSF to PSS). Mg++ (12 or 24 mM) was added to the system to determine the protective effect. The PSS was changed when oxygen saturation f ell to 5% in order to maintain respiration up to 5 hours. Results are expressed as mmoles of oxygen/min/g dw. Various vasoactive agents and modulators of intracellular signalling were added to the medium with SAH CSF.
Force Measurements
Isometric force measurements were performed on the porcine carotid artery. Maximal tension was generated with 70 mM KCl and is referred to has maximal tension. CSF from vasospasm patients and non-vasospastic controls was added to the aerated chambers a nd the tension responses recorded. Mg++ (12 or 24 mM) was added to the system to determine the protective effect.
Results
Oxygen consumption rate experiments:
Control experiments demonstrate that the porcine carotid artery could maintain a steady state, basal oxygen consumption rate (0.23±0.03 mmol of oxygen/min/g dw) in buffer for 5 hours. This rate was significantly decreased by 12 mM Mg++ (Figure 1) There was no difference in the percent water from all groups (75.1±1.4% and 74.8±1.7 respectively; N=12).
Addition of SAH CSF (1:30 dilution) from patients with vasospasm (SAH CSFv) stimulated the oxygen consumption rate in resting porcine carotid from contr ol levels to a new steady state (1.087±0.19 mmol of oxygen/min/g dw) after 300 minutes (Figure 1). Multiple rinses of the buffer medium had no effect on the acceleration of oxygen consumption. Patients without angiographic evidence of vasospasm (SAH CSFc) failed to stimulate oxygen consumption rate, as did "normal" CSF or extradural or subdural haematoma. Qualitatively similar results were seen with serum samples from these patients suggesting that the vasoactive factor is getting into the blood stream.
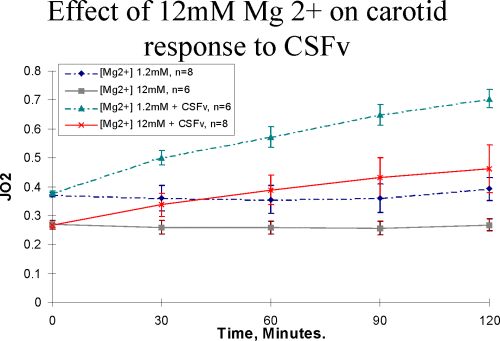 figure 1
figure 1
As seen in Table 1, Mg++ significantly decreased the tension generated by the tissue.
TABLE 1. Solution Conditions % Maximal Tension Maximal Tension (with KCl) 100 +/- SEM CSF Tension 124.9+/- 12 12 mM Mg++ administration 86.0 +/- 13 24 mM Mg++ administration 52.5 +/- 7 Pre-treat with 12 mM Mg++ 58.2 +/- 15
Discussion and Conclusion
Vasospasm accounts for considerable mortality and morbidity in a variety of pathological conditions afflicting the heart, bowel and brain. Cerebral vasospasm following subarachnoid haemorrhage occurs in 50-60% of the patients surviving SAH. It becomes sym ptomatic in 40% and causes significant mortality and morbidity in this patient population. The failure of powerful vasoactive agents to influence SAH cerebral vasospasm in the laboratory (Varsos et al., 1983) or in clinical practice has lead to two broad areas of research; in that structural versus metabolic mechanisms may be the "final common" pathway to vasospasm. The clinical observations of the eventual (more than 2 weeks) reversibility of lumenal narrowing (Weir et al., 1978) suggests that the vascul ar pathology may be metabolic (Butler, et al., 1996) though structural changes have been seen in early post-mortem studies (Hughes and Schianchi, 1978). The results presented here support a metabolic mechanism leading to vasospasm which may reversed or de creased by Mg++ administration.
ADP has been shown to produce a vasospastic like state in permeabilised VSM; slowing its ability to relax (Nishiye et al., 1993 and Fuglsang et al., 1993). This is because intracellular ADP has a relatively high affinity for myosin ATPase (Clark, 1994; Clark et al., 1994). Not surprisingly, therefore, the concentration of intracellular ADP is well controlled under most physiologic conditions (Clark, et al., 1995).
Above 60 mmol/l myosin ATPase will become progressively inhibited and VSM will fail to relax normally (Nishiye et al., 1993 and Fuglsang et al., 1993). Mitochondria will however become progressively stimulated at these ADP levels. The result is that intact mitochondria will consume oxygen, via oxidative phosp horylation, and maintain a low (50 umol/l) ADP. Uncoupled mitochondria will however, consume large quantities of oxygen but not replace the ATP consumed. We felt it necessary to determine if cerebral vasospasm would be a suitable model to examine the acti on(s) of ADP on smooth muscle metabolism and also to verify normal function in coupled mitochondria. Therefore the metabolic and functional changes seen during cerebral vasospasm may have a common pathological origin.
References
1) Clark, J.F. The Creatine Kinase System in Smooth Muscle. Molecular And Cellular Biochem. 133/134, 221-232, 1994.
2) Clark, J.F., G.J. Kemp and G.K. Radda. The Creatine Kinase Equilibrium Free [ADP] and Myosin ATPase in Vascular Smooth Muscle Cross-Bridges. Journal of Theoretical Biology. 173, 207-211, 1995.
3) Clark, J.F., Z. Khuchua, A.V. Kuznetsov, E.A. Boehm and R. Ventura-Clapier. Creatine Kinase Activity Associated with the Contractile Proteins of the Guinea Pig Carotid Artery. Journal of Muscle Research and Cell Motility. 15, 432-439, 1994.
4) Clark, J.F., Z. Khuchua, A.V. Kuznetsov, V.A. Saks, and R. Ventura-Clapier. The compartmentation and Role of Creatine Kinase in the Myometrium of the Gestating Guinea Pig Uterus. Journal of Physiology, (London), 466, 553-572, 1993.
5) Fuglsang, A., A. Khromov, K. Torok, A.V. Somlyo, and A.P. Somlyo. Flash Photolysis Studies of Relaxation and Cross-Bridge Detachment: Higher Sensitivity of Tonic than Phasic Smooth Muscle to MgADP. Journal of Muscle Research and Cell Motility. 14, 6 66-673, 1993.
6) Hughes, J.T., and P.M. Schianchi. Cerebral Artery Spasm. A histological Study at Necropsy of the Blood Vessels in Cases of Subarachnoid Hemorrhage. Journal of Neurosurgery. 48, 515-525, 1978.
7) Kim P. Loss of Relaxations Metabolic Failure and Increased Calcium Permeability of Smooth Muscle During Chronic Cerebral Vasospasm. J. Autonomic Nervous System. 49, S157-S162, 1994.
8) Kim, J-C, B. Wier, R.L. MacDonald, L.S. Marton, H. Zhang. Hemolysate Inhibits L-Type Ca++ Channels in Rat Basilar Smooth Muscle Cells. Journal of Vascular Research. 33, 258-264, 1996.
9) Kim, P., J.D. Jones, and T.M. Sundt. High Energy Phosphate Levels in the Cerebral Artery During Chronic Vasospasm after Subarachnoid Hemorrhage. Journal of Neurosurg. 76, 991-996, 1992.
10) MacDonald R.L., and Bryce K.A. Weir. A Review of Haemoglobin and the Pathogenesis of Cerebral Vasospasm. Stroke 22, 971-982, 1991.
11) Nishiye, E., A.V. Somlyo, K. Torok, and A.P. Somlyo. The Effects of MgADP on Cross-Bridge Kinetics: a Laser Flash Photolysis Study of Guinea Pig Smooth Muscle. Journal of Physiology. (London) 460, 247-271, 1993.
12) Petrok, K.C., West, M. Mohr A. Nimodopine Treatment in Poor Grade Aneurysm Patieints. Journal of Neurosurgery, 68, 505-517, 1988.
13) Pichard, J.D. A.P, Murray, Illingworth, R et al., Effect of Oral Nimodopine on Cerebral Infarction and Outcome after Subarachnoid Haemorrhage. British Medican Journal, 298, 636-642, 1989.
14) Symon, L. Disordered Cerebrovascular Physiology in Aneurysmal Subarachnoid Haemorrhage. Acta Neurochim. (Vienna) 41, 7-22, 1978.
15) Varsos, V.G, T.M. Liszczak, D.H. Han, J.P. Kistler, J. Vielma, P.M. Black, R.C. Heeros, N.T. Zervas. Delayed Cerebral Vasospasm is not Reversible by Aminophylin, Nifedepine or Papaverine in ‘Two-Hemorrhage’ Canine Model. J. Neurosurg. 58, 11-17, 19 83.
16) Weir B. The Pathophysiology of Cerebral Vasospasm. British Journal of Neurosurgery. 9, 375-390, 1995.
17) Weir, B., M. Grace, Havsen, J. Rothberg, Time Course of Vasospasm in Man. Journal of Neurosurgery. 48, 173-178, 1978.
| Discussion Board | Previous Page | Your Symposium |