Invited Symposium: SERCA-Type of Calcium Pumps and Phospholamban
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Phospholamban (PLB) is a low molecular weight phosphoprotein associated with cardiac sarcoplasmic reticulum (SR). The dephosphorylated form of PLB is an inhibitor of the SR Ca2+-ATPase affinity for Ca2+. Phosphorylation of PLB relieves its inhibitory effects on the SR Ca2+ pump, with subsequent acceleration of Ca2+ transport into the SR lumen, which is thought to underlie the positive inotropic and lusitropic actions of beta-adrenergic agonists in the mammalian heart. The role of PLB in the regulation of basal myocardial contractility has been recently elucidated through the development of a PLB knockout (KO) mouse via gene-targeting methodology in embryonic stem cells. The PLB knockout mouse hearts exhibited significant increases in the SR Ca2+-pump affinity for Ca2+ and in the basal contractile parameters compared with wild-type littermates. The cardiac phenotype was analyzed utilizing an integrative approach and combining studies at the subcellular, organ and whole animal levels. Furthermore, the inotropic and lusitropic effects of beta-agonists on cardiac function were significantly attenuated in PLB knockout mice compared with wild-type mice. These studies indicated that PLB is a critical inhibitor of basal cardiac function and a major regulator of the heart's responses to beta-adrenergic agonists.
Phospholamban was originally proposed to form homopentamers through interactions between its transmembrane domain in the SR, based on its migration pattern in SDS-polyacrylamide gels. Recently, alanine-scanning mutagenesis studies in expression systems showed that a monomeric form of PLB, in which Leu-37 was changed to Ala, suppressed SR Ca2+-ATPase Ca2+ affinity more effectively than wild-type or pentameric PLB. It was thus postulated that there might be an equilibrium between pentameric and monomeric PLB in SR membranes. Pentameric PLB may represent a less active reservoir, which dissociates to provide active monomeric PLB subunits. However, co-expression of other monomeric forms of PLB, associated with mutation of Cys-41 to Phe or Ser, with SR Ca2+-ATPase in HEK-293 cells failed to demonstrate super-inhibition (or gain of function) by PLB monomers. Furthermore, recent studies indicated that a pentameric form of PLB, resulting from mutation of Asn-27 to Ala was more potent than a monomeric mutant (L37A) PLB in inhibiting the SR Ca2+-ATPase Ca2+ affinity, and suggested that the oligomeric structure of PLB may be important in SR Ca2+-ATPase regulation.
The availability of the PLB knockout mouse in conjunction with site-specific mutagenesis technology has provided us with an excellent opportunity to examine structure-function relations of PLB in a background free of the endogenous protein. In this article, we will describe the feasibility of reinsertion of the PLB gene in the PLB knockout heart and present evidence for the functional unit (pentamers vs. monomers) of PLB in vivo.
Phospholamban Ablation and Hyperdynamic Cardiac Function
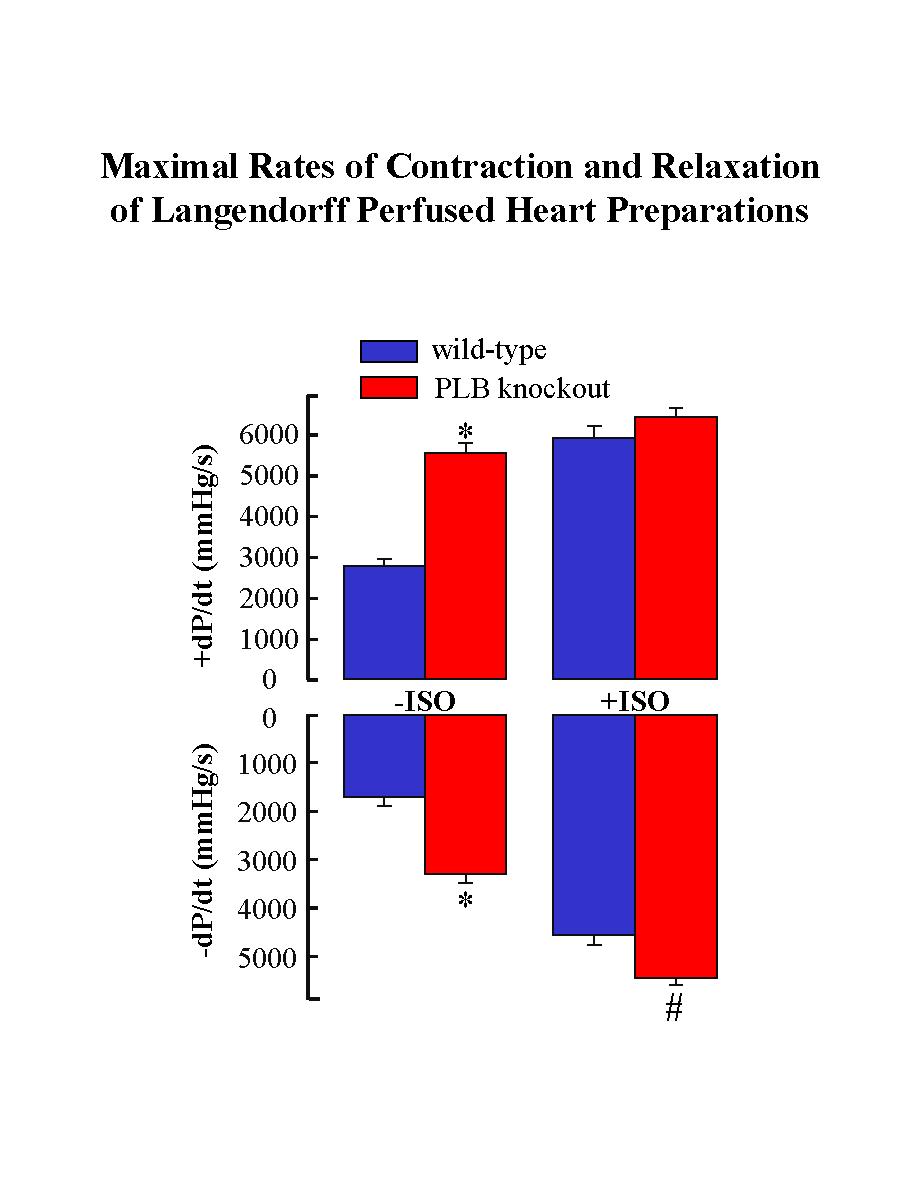
The mutant mice deficient in PLB were generated by gene-targeting methodology in embryonic stem cells. Phospholamban ablation was not associated with any phenotypic changes at the gross level. Examination of the hearts at the ultrastructural level revealed no cytoarchitectural disruption in the PLB knockout mice compared to age-matched wild-type mice. The ultrastructure, location and organization of the SR, the subcellular membrane system known to contain phospholamban, were identical in the cardiac tissue from wild-type and PLB knockout mice. However, Langendorff perfusion of PLB knockout hearts indicated significant enhancement of the myocardial contractile parameters (Fig. 1) with no changes in the spontaneous heart rate.
 Click to enlarge
Fig. 1: The wild type and PLB knockout hearts were perfused in the absence (-) and presence (+) of isoproterenol (ISO). * P < 0.05 vs. wild type without ISO; # P < 0.05 vs. PLB knockout with ISO.
Click to enlarge
Fig. 1: The wild type and PLB knockout hearts were perfused in the absence (-) and presence (+) of isoproterenol (ISO). * P < 0.05 vs. wild type without ISO; # P < 0.05 vs. PLB knockout with ISO.
The time to peak pressure (TTP) and half-relaxation time (RT1/2) were also significantly shortened by PLB deficiency. Maximal isoproterenol perfusion was associated with significant stimulation in the rates of contraction (+dP/dt: 208%) and relaxation (-dP/dt: 260%) in the wild-type hearts, whereas it only slightly stimulated the relaxation rate in the PLB knockout hearts (Fig. 1).
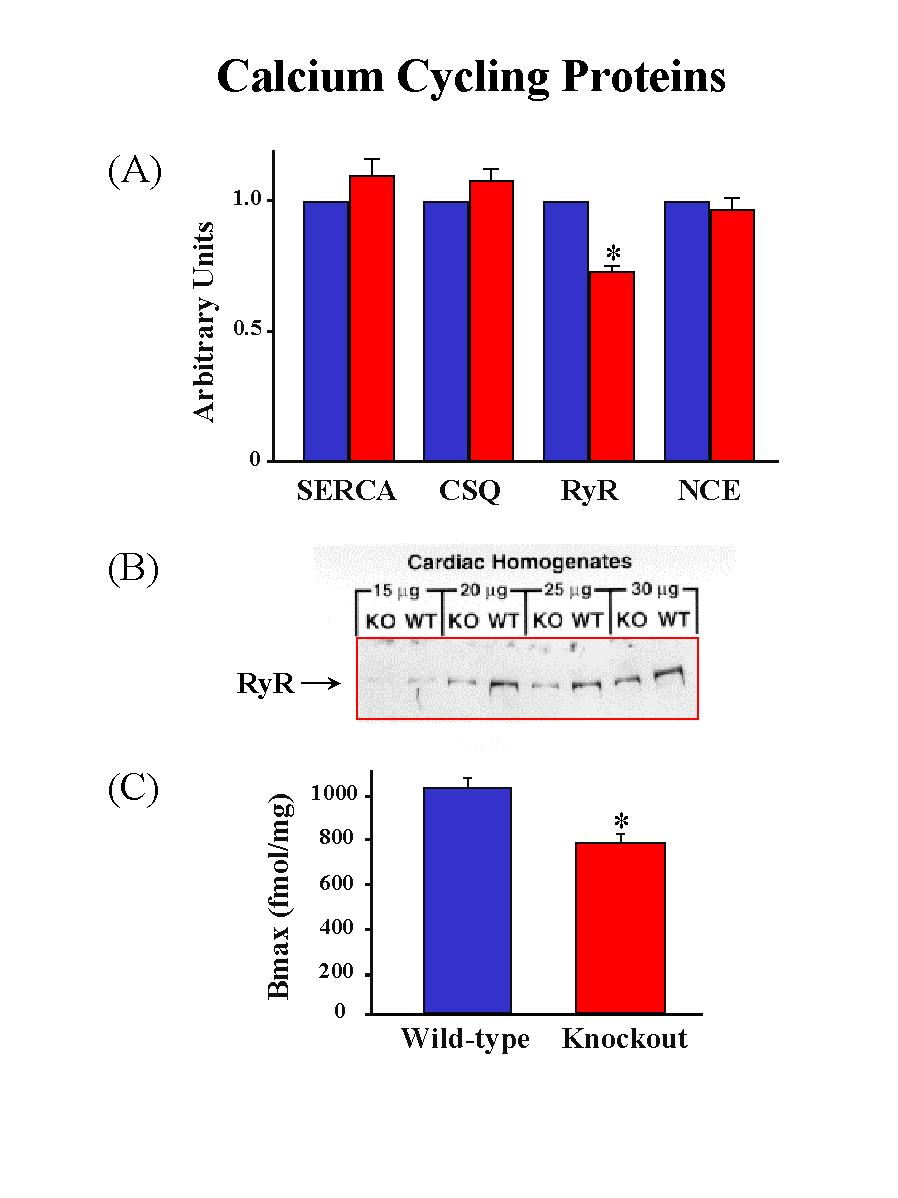
To determine whether ablation of PLB and the accompanied enhancement of cardiac contractile parameters were associated with any alterations of the SR Ca2+-ATPase, the activity of this enzyme was examined in Ca2+-uptake experiments. The initial rates of the SR Ca2+ uptake in cardiac homogenates were assessed as a function of [Ca2+]. There was no alteration in the Vmaxof the SR Ca2+ transport system, indicating that the expression levels and the activity of the SR Ca2+-ATPase were not altered in the PLB knockout hearts. However, the affinity of the SR Ca2+-ATPase for Ca2+, a property modulated by PLB, was increased upon ablation of PLB. Furthermore, dot blot analysis and quantitative immunoblotting revealed no differences in either the transcript or the protein levels of the SR Ca2+-ATPase between wild-type and PLB knockout hearts (Fig. 2A).
 Click to enlarge
Fig. 2: Quantification of the major calcium cycling proteins. SERCAS: SR Ca2+-ATPase; CSQ: calsequestrin; RyR: ryanodine receptor; NEC: Na+-Ca2+ exchanger.
Click to enlarge
Fig. 2: Quantification of the major calcium cycling proteins. SERCAS: SR Ca2+-ATPase; CSQ: calsequestrin; RyR: ryanodine receptor; NEC: Na+-Ca2+ exchanger.
Examination of the transcript or the protein levels of calsequestrin and the sarcolemmal Na+-Ca2+ exchanger indicated no significant differences between PLB knockout and wild-type hearts. However, phospholamban deficiency was associated with a significant reduction (26%) in the levels of the ryanodine receptor protein (Fig. 2B). The reduction in protein levels was consistent with a 24% decrease in ryanodine binding in PLB knockout hearts compared with wild-type hearts (Fig. 2C), while the ryanodine binding affinity was similar between PLB knockout and wild-type hearts.
The contractile proteins, myosin, actin and troponins are known to play a major role in controlling the muscle contractile machinery. Thus, the transcript levels of these proteins were evaluated by dot blot analysis. There were no significant alterations in the mRNA levels of alpha-myosin heavy chain, alpha-cardiac actin, alpha-skeletal actin, or troponin I between PLB knockout and wild-type hearts. Furthermore, there were no alterations in the protein levels of myosin, actin, troponin T and troponin I. Examination of beta-myosin heavy chain expression indicated that these transcript levels were undetectable in PLB knockout or wild-type hearts, suggesting that PLB ablation did not trigger any transition between alpha- and beta-myosin heavy chain proteins in an effort to accommodate the enhanced SR Ca2+-ATPase activity and hyperdynamic cardiac function in the intact mouse heart.
Reinsertion of Phospholamban into the Knockout Mouse Heart
To determine whether the hyperdynamic cardiac function of the PLB knockout mouse can be reversed by reintroduction of the missing PLB gene, we used the alpha-myosin heavy chain (alpha-MHC) promoter to direct expression of mouse wild-type PLB cDNA in the cardiac compartment of the PLB knockout mouse (KO+WT). Four KO+WT transgenic lines were identified by PCR and Southern blot analyses of mouse genomic DNA. Northern blot analysis of total RNA isolated from the hearts of the KO+WT transgenic mice revealed expression of the transgene. To assess the protein levels of PLB in the transgenic hearts, we performed quantitative Western blot analysis of cardiac homogenates from KO+WT in parallel with wild-type control mice. The line, which expressed PLB levels (0.7-fold) closer to those present in control hearts, was chosen for evaluation of the physiological effects of PLB reintroduction in the knockout background. Sarcoplasmic reticulum membranes isolated from the transgenic hearts indicated that the reintroduced PLB was inserted in the SR and there were no alterations in the SR Ca2+-ATPase levels compared to control hearts.
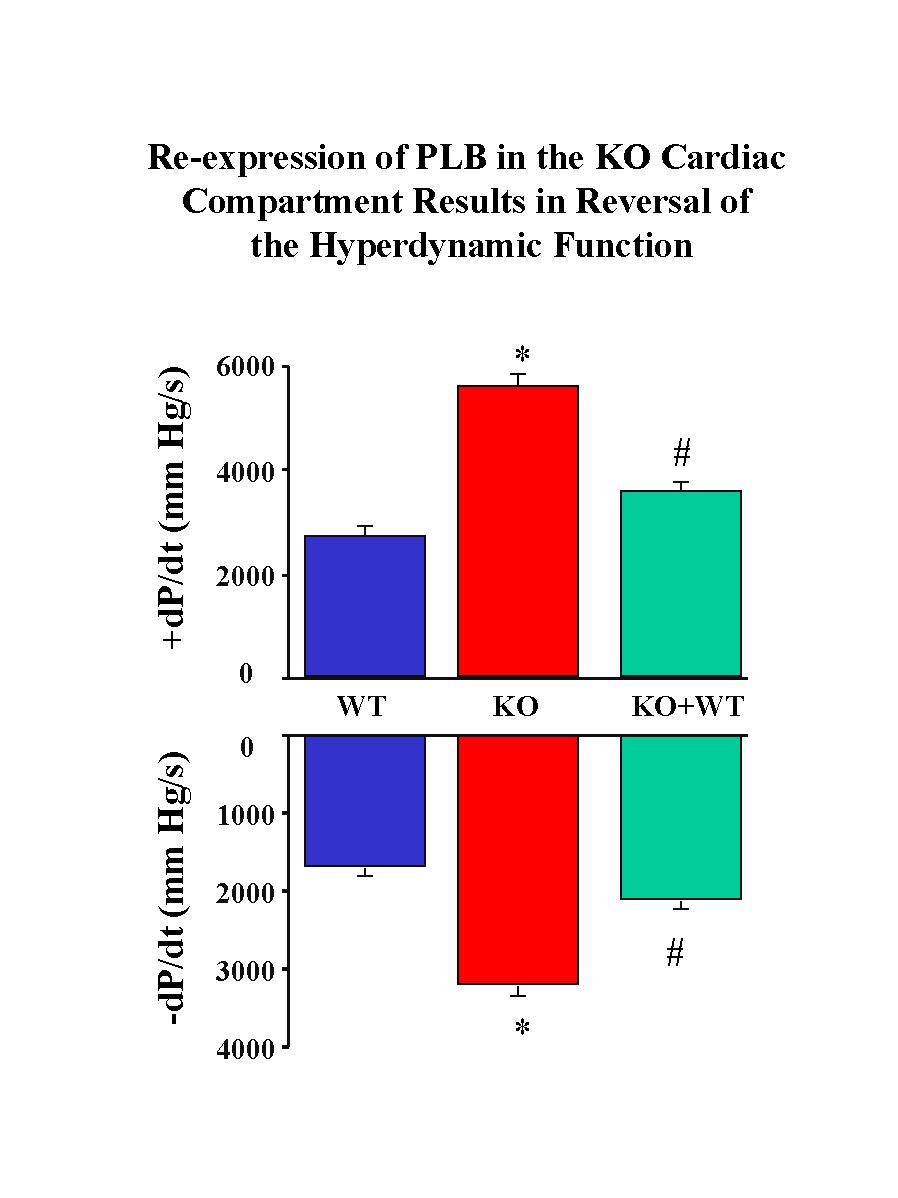
To determine whether the reintroduced PLB was capable of reversing the hyperdynamic cardiac function associated with PLB deficiency, hearts from KO+WT and PLB knockout mice were subjected to Langendorff perfusion in parallel with control hearts. The PLB knockout hearts exhibited significantly enhanced myocardial performance compared to wild-type controls, as characterized by significant increases in the maximal rates of cardiac contraction and relaxation (�dP/dt) (Fig.3).
 Click to enlarge
Fig. 3: Langendorff-perfused heart preparations. WT: wild type control; KO: PLB knockout hearts; KO+WT: reintroduction of PLB in the knockout mouse hearts.
Click to enlarge
Fig. 3: Langendorff-perfused heart preparations. WT: wild type control; KO: PLB knockout hearts; KO+WT: reintroduction of PLB in the knockout mouse hearts.
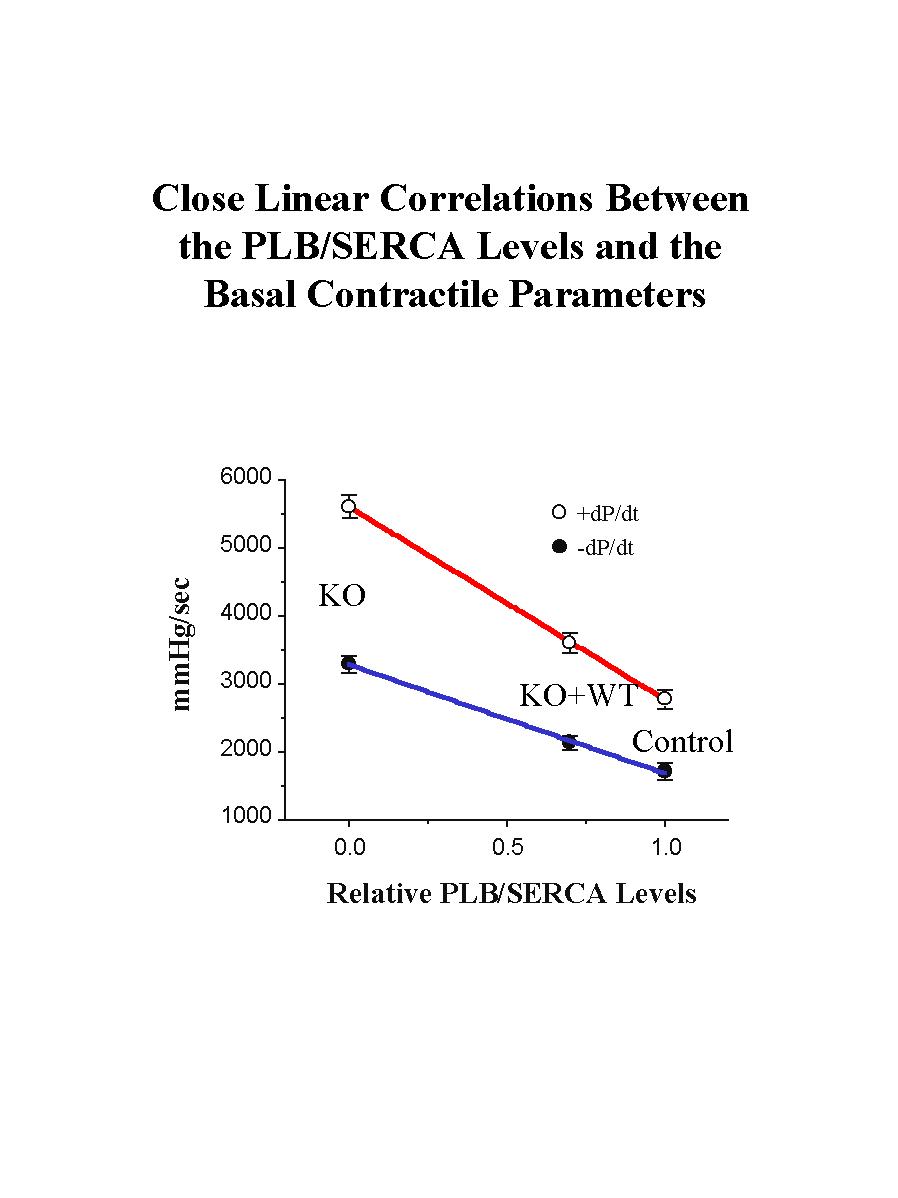
Reinsertion of PLB in the knockout background was associated with significant depression of the contractile parameters (Fig. 3). Please note that reversal of the enhanced cardiac contractile parameters was not complete, or to the levels observed in control hearts, since the levels of reintroduced PLB were 0.7-fold of those present in control hearts. Interestingly, when the relative levels of PLB or PLB/SR Ca2+-pump in the three animal models were plotted against the maximal basal rates of contraction (+dP/dt) and relaxation (-dP/dt), a close linear correlation was observed (Fig. 4), consistent with our previous observations in PLB knockout, PLB-heterozygous and control hearts.
 Click to enlarge
Fig. 4: The relative ratios of PLB to the SR Ca2+-ATPase (SERCA) were plotted against the basal maximal rates of contraction (+dP/dt) and relaxation (-dP/dt) in the perfused hearts from various mouse models: wild type control (control), PLB knockout (KO), and transgenic mice expressing the wild type PLB in the null background (KO+WT).
Click to enlarge
Fig. 4: The relative ratios of PLB to the SR Ca2+-ATPase (SERCA) were plotted against the basal maximal rates of contraction (+dP/dt) and relaxation (-dP/dt) in the perfused hearts from various mouse models: wild type control (control), PLB knockout (KO), and transgenic mice expressing the wild type PLB in the null background (KO+WT).
These findings indicate that the hyperdynamic PLB knockout phenotype can be reversed by reintroduction of PLB in the null background, and demonstrate the potential power of this technology in performing structure-function studies of PLB in vivo.
Functional Unit of Phospholamban In Vivo
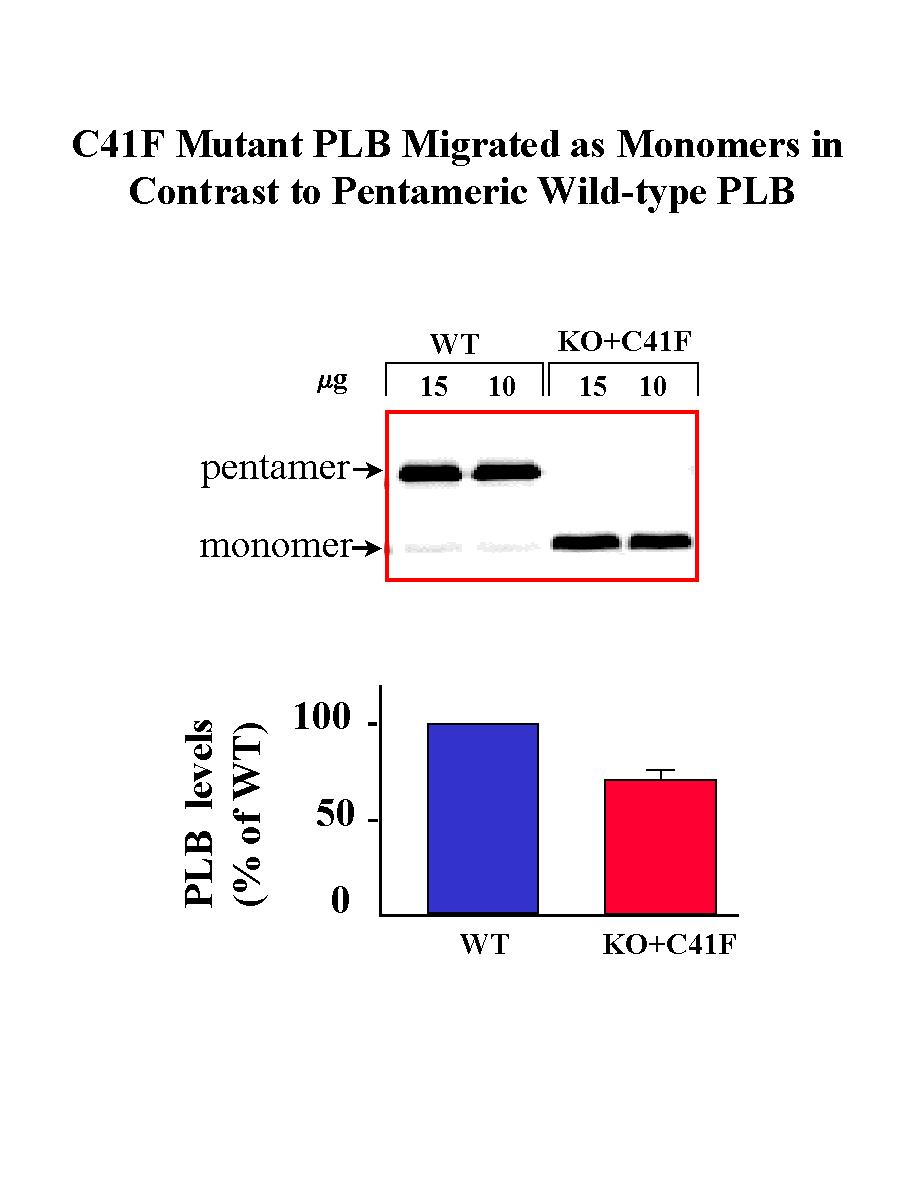
The reversal of the hyperdynamic contractile parameters of the PLB knockout hearts by reintroduction of the wild-type PLB demonstrated the feasibility of reinserting various PLB mutants in the knockout background and assessing their functional relevance in vivo. Thus, to better clarify the physiological effects of monomeric PLB, mutation of Cys-41 to Phe in PLB was performed by PCR site-directed mutagenesis, and expression of the mutated PLB was driven by the alpha-MHC promoter in the PLB knockout mice (KO+C41F), in an identical manner as wild-type PLB described above. The C41F mutation was chosen based on previous in vitro and in vivo studies, which indicated that this mutation resulted in pentamer destabilization and formation of PLB monomers. The transgene and its expression were detected using Southern, Northern and Western blot analyses in the transgenic lines, as previously described. As expected, the C41F mutant PLB migrated on SDS-gels as monomers whereas the wild-type PLB mainly migrated as pentamers (Fig. 5).
 Click to enlarge
Fig. 5: Quantitative immunoblotting of PLB in wild type control (WT) and transgenic mice expressing C41F mutant PLB in the PLB knockout background (KO+C41F).
Click to enlarge
Fig. 5: Quantitative immunoblotting of PLB in wild type control (WT) and transgenic mice expressing C41F mutant PLB in the PLB knockout background (KO+C41F).
One of the lines expressing 0.7-fold PLB, which was similar to the PLB levels expressed in KO+WT hearts, was selected for breeding and further studies. Analysis of the SERCA levels showed that there was no alteration upon introduction of mutant PLB in the mouse heart.
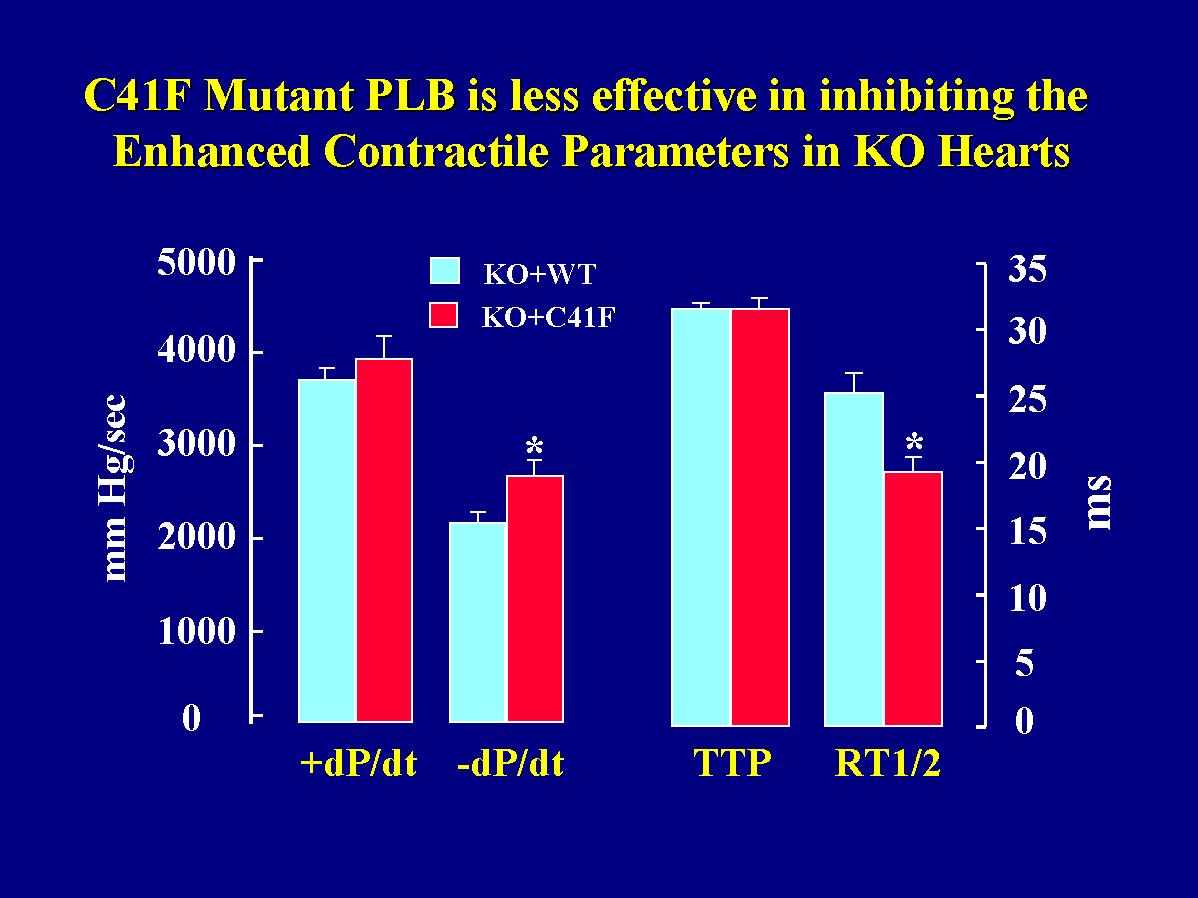
As described above, the rates of cardiac contraction and relaxation were significantly increased upon ablation of PLB compared to wild-type hearts. Conversely, reintroduction of wild-type PLB in the knockout hearts reversed their hyperdynamic function. To determine whether reintroduction of monomeric PLB had similar regulatory effects as wild-type or pentameric PLB, the hearts from transgenic mice with wild-type or mutant PLB reintroduced into the knockout background were perfused in parallel in a retrograde mode. Langendorff-perfusion indicated that there was no difference in +dP/dt values between the two transgenic groups, but the -dP/dt values were depressed to a greater extent by reintroduction of wild-type PLB than mutant PLB in the knockout hearts (Fig. 6).
 Click to enlarge
Fig. 6: Effects of the C41F mutant PLB on contractile parameters in Langendorff-perfused hearts. +dP/dt and -dP/dt: the maximal rates of contraction and relaxation; TTP: time to peak pressure; RT1/2: time to half relaxation.
Click to enlarge
Fig. 6: Effects of the C41F mutant PLB on contractile parameters in Langendorff-perfused hearts. +dP/dt and -dP/dt: the maximal rates of contraction and relaxation; TTP: time to peak pressure; RT1/2: time to half relaxation.
This observation was also consistent with the time parameters in the same cardiac preparations. There was no difference in the time to peak pressure (TTP), while the time to half-relaxation (RT1/2) was prolonged significantly more in wild-type PLB than in mutant PLB hearts (Fig. 6). These findings indicate that the mutant monomeric PLB was not capable of depressing the enhanced relaxation parameters of the PLB knockout hearts to the same extent as wild-type PLB.
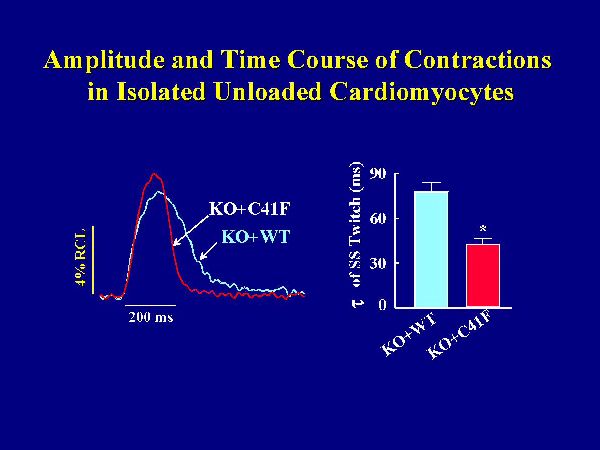
The hypothesis that monomeric PLB is less effective in inhibiting the cardiac contractility was further tested at cellular levels, using isolated cardiomyocytes. Left ventricular myocytes were isolated from KO+C41F and KO+WT transgenic mice, paced at 0.5Hz and used for measurements of contractile parameters and Ca2+ transients. Reintroduction of wild-type PLB in the knockout background was associated with significant depression of the cell shortening fraction and prolongation of the time constant of relaxation, compared to PLB knockout cardiomyocytes. Figure 7 shows representative traces of steady-state cell shortening of transgenic myocytes with wild-type PLB or mutant PLB reintroduced in the knockout background.
 Click to enlarge
Fig. 7: Myocyte twitch contractions. Left ventricular myocytes were isolated and paced at 0.5 Hz. All measurements were obtained during steady-state (SS) twitches at 1 mM Ca2+ and room temperature. KO+WT and KO+C41F: transgenic mice with wild type or C41F mutant PLB reintroduced in the knockout background.
Click to enlarge
Fig. 7: Myocyte twitch contractions. Left ventricular myocytes were isolated and paced at 0.5 Hz. All measurements were obtained during steady-state (SS) twitches at 1 mM Ca2+ and room temperature. KO+WT and KO+C41F: transgenic mice with wild type or C41F mutant PLB reintroduced in the knockout background.
Myocytes expressing wild-type PLB relaxed slower than those expressing mutant PLB. The time constant for cell relaxation during the steady-state twitch was significantly faster in the mutant PLB myocytes. The amplitude of cell shortening was slightly but not significantly higher in mutant PLB myocytes. Consistent with cell mechanics, the decline of the Ca2+ transient was faster in myocytes with mutant PLB than wild-type PLB. Peak [Ca2+]i tended to be higher with the mutant PLB, but again this effect was not significant. Thus, the time constants of relaxation and Ca2+ transient decline in isolated cardiomyocytes were diminished to a greater extent by wild-type than mutant phospholamban, even without significant differences in the amplitudes of myocyte contraction and Ca2+ transient between the two groups.
Summary and Conclusions
In summary, phospholamban ablation was associated with significant enhancement of the rates of contraction and relaxation, as well as attenuation of responses to beta-adrenergic stimulation in the mouse heart. The hyperdynamic cardiac function of the PLB knockout mice was not accompanied by any cytoarchitectural abnormalities or alterations in the expression levels of the SR Ca2+-ATPase, calsequestrin, Na+-Ca2+ exchange or the contractile proteins. However, ablation of phospholamban was associated with downregulation of the ryanodine receptor, to compensate for the increased SR Ca2+-uptake in an attempt to maintain Ca2+ homeostasis in the myocardium. Furthermore, the attenuation of the contractile responses to beta-agonists was not due to alterations in the phosphorylation levels of the other key cardiac phosphoproteins in the PLB knockout hearts. Thus, the augmentation of contractile parameters and the attenuation of beta-adrenergic stimulation in the PLB knockout hearts are mainly due to the "uninhibited" SR Ca2+ -ATPase activity as a result of PLB ablation.
The hyperdynamic cardiac phenotype in PLB knockout mouse can be reversed by reintroducing wild-type PLB into the PLB knockout heart. The alpha-myosin heavy chain promoter was used to direct cardiac-specific expression of wild-type PLB in the knockout background, and reversal of the hyperdynamic cardiac phenotype of the PLB-deficient hearts was observed. The degree of inhibition of the contractile parameters was proportional to the expression levels of PLB, in agreement with our previous studies in PLB-heterozygous and PLB-homozygous mice. The success of PLB transgenesis in the genetically altered background, accompanied by reversal of the knockout phenotype, indicated that the PLB-deficient mouse provides an attractive model system for expression of various PLB mutants in the heart and elucidation of structure-function relationships in vivo. Thus, we reintroduced wild-type (pentameric) or monomeric mutant (C41F) PLB in the hearts of PLB knockout mice, and characterized their cardiac phenotypes in parallel. Expression of a monomeric mutant PLB in which Cys-41 was replaced by Phe in the knockout background indicated that the cardiac relaxation and Ca2+ transient decline in isolated cardiomyocytes were diminished to a greater extent by wild-type than mutant PLB. Langendorff perfusion also indicated that the mutant PLB was not capable of depressing the enhanced relaxation parameters of the PLB knockout hearts to the same extent as wild-type PLB. Thus, the mutant or monomeric form of PLB was not as effective in slowing Ca2+ decline or relaxation in cardiomyocytes or perfused hearts as wild-type or pentameric PLB. These findings suggest that pentameric assembly of PLB may be necessary for optimal regulation of myocardial contractility in vivo.
REFERENCES
- Luo, W., Grupp, I. L., Harrer, J., Ponniah, S., Grupp, G., Duffy, J. J., Doetschman, T., and Kranias, E. G. (1994) Circ. Res. 75, 401-409.
- Koss, K.L., and Kranias, E.G. (1996) Circ. Res. 79, 1059-1063.
- Chu, G., Dorn II, G.W., Luo, W., Harrer, J.M., Kadambi, V.J., Walsh, R.A., and Kranias, E.G. (1997) Circ. Res. 81, 485-492.
- Luo, W., Chu, G., Sato, Y., Zhou, Z., Kadambi, V.J., and Kranias, E.G. (1998) J. Biol. Chem. 273, 4734-4739.
- Wegener, A.D., and Jones, L.R. (1984) J. Biol. Chem. 259, 1834-1841.
- Colyer, J. (1993) Cardiovasc. Res. 27, 1766-1771.
- Kovacs, R.J., Nelson, M.T., Simmerman, H.K.B., and Jones, L.R. (1988) J. Biol. Chem. 263, 18364-18368.
- Chu, G., Li, L., Sato, Y., Harrer, J.M., Kadambi, V.J., Hoit, B.D., Bers, D.M., and Kranias, E.G. (1998) J. Biol. Chem. 273(50), 33674 (in press).
- Fujii, J., Maruyama, K., Tada, M., and MacLennan, D.H. (1989) J. Biol. Chem. 264, 12950-12955.
- Cornea, R.L., Jones, L.R., Autry, J.M., and Thomas D.D. (1997) Biochemistry 36, 2960-2967.
- Autry, J.M., and Jones, L.R. (1997) J. Biol. Chem. 272, 15872-15880.
- Kimura, Y., Kurzydlowski, K., Tada, M., and MacLennan, D.H. (1997) J. Biol. Chem. 272, 15061-15064.
- Toyofuku, T., Kurzydlowski, K., Tada, M., and MacLennan, D.H. (1994) J. Biol. Chem. 269, 3088-3094.
- Kimura, Y., Asahi, M., Kurzydlowski, K., Tada, M., and MacLennan, D.H. (1998) J. Biol. Chem. 273, 14238-14241.
- Kiss, E., Edes, I., Sato, Y., Luo, W., Liggett, S.B., and Kranias, E.G. (1997) Am. J. Physiol . 272, H785-H790.
- Karim, C.B., Stamm, J.D., Karim, J., Jones, L.R., and Thomas, D.D. (1998) Biochemistry 37, 12074-12081.
| Discussion Board | Previous Page | Your Symposium |