Invited Symposium: Photodynamic Therapy
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Each year, an incidence of 15,000 new cases of primary confirmed brain tumours is reported in the U.S. alone. The incidence rate appears to increase with age up to 65 years, with the most frequent brain tumours being glioblastoma multiform (29%), meningiomas (18%), anaplastic astrocytomas (11%), and astrocytomas (9%) [16]. In spite of surgical tumour resection and vigorous adjuvant radiotherapy and/or chemotherapy, the prognosis for these patients is poor, with a 1.8-year mean survival period from the time of diagnosis noted for all cases [16].
Intraoperative treatment of malignant brain tumours by Photodynamic Therapy (PDT) using various photosensitizers, most notably, Photofrin, has been investigated in several clinical trials with some evidence of increased survival [18, 27]. In a previous study [14] we quantified tissue response to PDT-mediated by four different photosensitizers based on the Photodynamic threshold model [28] using coagulative tissue necrosis as biological end point. This study showed marked differences in bulk photosensitiser uptake and PDT response, for the different tissue-photosensitizers combinations. Thus, not only the sensitivity of the tumour to PDT but also that of the surrounding normal brain tissue need to be considered when administering intracranial PDT. For examples, it was noted that very infrequent and only very small lesions were observed in white mater following aminolevulinic acid (ALA) induced protoporphyrin IX (PPIX) mediated PDT, while the cortex and VX2 tumour are very sensitive. Hence, ALA induced PPIX may be the photosensitiser of choice for tumours situated in the frontal lobes, which are mostly embedded in white matter.
While coagulative necrosis is generally the accepted mode of brain cell death induced by intracranial PDT, apoptosis, or programmed cell death, has not yet been investigated. Coagulative necrosis results from direct physical injury to the cell or from destruction of the vasculature and subsequent ischemic hypoxia, apoptosis is considered a deliberate response to specific, and possibly indirect stimuli [7, 24]. Yoshida et al. [27] report that responses of the brain to PDT can extend beyond the cells that are initially killed by coagulative necrosis and that structural signs of lethal injury to neurones in brain adjacent to tumour (BAT) develop over a period of 18 hours after PDT.
For ionising radiation, numerous clinical studies reported that following irradiation of the central nervous system (CNS) a significant portion of patients develops complications, after a latent period of months to years. These include demyelination of nerve fibres and white matter necrosis, leading to devastating neurological disorders [9, 22, 23, 26]. Currently the apoptotic response of the CNS to ionising radiation, as one possible source for the latent complications is currently under intense investigation [10, 11, 12]. The effects of PDT administered to the brain and CNS have not been completely characterised. Previous investigations have observed PDT-induced apoptosis in vitro [8, 20] and have reported that in vivo, PDT-induced apoptosis is an early event, occurring completely within the first 24 hours after treatment [27].
As apoptosis can occur in regions remote from the treatment site (e.g. the tumour) it could contribute to the neurological deficits associated with PDT [18]. Apoptosis of glial cells can lead to demyelination of nerve fibres and subsequently to reductions in the efficiency of neural conduction. Hence, the clinical implications of PDT-induced apoptosis in the brain are significant. The present study, is a preliminary retrospective examination of the PDT-induced apoptotic response in normal rabbit brain and VX2 tumour 24 hours post PDT mediated by 5 different photosensitizers as used in the previous study [14]. Originally the experiments were designed to investigate the tissue response to PDT using the photodynamic threshold model and thus this can only be a preliminary study to determine the presence of apoptotic cells or bodies at this single time point. If different photosensitizers produce distinct apoptotic effects in normal brain, BAT, or the VX2 tumour, it may be possible to choose a particular photosensitiser to elicit a desired apoptotic response.
Materials and Methods
New Zealand White rabbits (male, 2-3.5kg) were used for the original study of PDT induced necrosis for four photosensitizers [14]. The rabbit model was selected initially to provide a large enough cranial cavity to allow for several mm large lesions without compromising the survival of the animal. Since no glial tumour model is available for the rabbit, a VX2 carcinoma (a virus-induced papilloma which grows as a metastatic brain tumour) was chosen. This tumour exhibits various characteristics of primary brain tumours, including microinvasion, pseudo-palisading, and growth along blood vessels and in perivascular spaces, causing breakdown of the blood-brain barrier (BBB) within the tumour and in BAT.
Animals were randomised for tumour implantation, photosensitiser (type, dose, and time delay before irradiation), or sham treatment, and several representative animals were chosen from these groups for this preliminary analysis, as listed in Table I. Six additional animals received meta-Tetra (hydroxyphenyl) chlorine (mTHPC) mediated PDT for the purposes of this study. Control animals received either light or photosensitiser alone. Since PDT was only performed on the left hemisphere of the brains, the contralateral hemispheres served as controls to indicate if administration of photosensitiser alone caused apoptosis.
The protocol is described in detail elsewhere [13,14] in detail. Briefly; Photofrin (porfimer sodium) was supplied as a freeze-dried powder (QLT, Vancouver, B.C., Canada) reconstituted to 5mg/mL in 5% dextrose solution. Aminolevulinic acid (ALA) was obtained in hydrochloride form (Sigma Scientific, St. Louis, MO, USA), dissolved in phosphate buffered saline (PBS, 80g/mL) and the pH was adjusted to 6.5-6.8 with 1N NaOH. Dr. Nancy Oleinick (Case Western Reserve University, Cleveland, OH, USA) supplied non-sulfonated chloroaluminum phthalocyanine (AlClPc) in an emulsion carrier (1mg/mL). Tin Ethyl Etiopurpurin (SnET2) (Purlytin; Miravant, Santa Barbara, CA, USA) was supplied ready for injection in two different delivery vehicles: emulsion at 1 mg/ml, and liposome at 0.87 mg/ml. mTHPC (Scotia Drug Discovery, London, UK) was reconstituted to 0.5mg/ml in chlorine diluent (30% polyethylene glycol 40, 20% ethanol, 50% distilled water: Scotia Pharma Ltd., London, UK). All photosensitizers were injected i.v. at a rate <1ml/min, 24 hours prior to PDT light-treatment, with the exception of ALA, which was administered six hours before light treatment.
For tumour implantation, a burr hole was drilled 5mm posterior to the sagital suture and 5mm left of the midline and 105 VX2 cells in 50�l PBS were injected 6mm below the dura. After a 13-day growth period, tumours of 8-12mm diameter were obtained. For PDT, a left parietal bone craniotomy (~12 x 8 mm) was performed and the dura was removed. PDT treatment light [~ 35 mW from an argon laser (Ion-Laser Technologies, Salt Lake City, UT, USA)] was delivered via a quartz optical fibre with a flat-cleaved end, to a depth of 6mm into the centre of the tumour or for non-tumour bearing animals at the same anatomical site. A wavelength of 514nm was used, since the high extinction coefficient of brain tissues at 514nm limits the light penetration to a few millimetres, thereby restricting the size of PDT-induced lesions and related oedema. Two animals administered with SnET2 (emulsion) were treated at 660 nm the absorption maximum of SnET2 resulting in large lesions and raised intracranial oedema and pressure. Following treatment, the animals were allowed to recover and an analgesic (0.2 ml Temgesic, Reckitt-Colman, Hull, UK s.c. every 8 h) was administered until the time of sacrifice, 24 hours post PDT by lethal injection (T-61: Hoechst, Regina, SK, Canada).
The brains were removed intact, fixed in 10% formalin in PBS for 10 days, and cut into 3mm-thick transverse slices. The slice displaying the largest diameter of necrosis was routinely processed, embedded in paraffin, and serial sections, of 4-6 �m thickness, were cut from it. The sections were then stained with fluorescein-12-dUTP* (deoxy-uridine-tri-phosphate) in a TdT (terminal deoxy nucleotide transferase) assay, using the Apoptosis Detection System, Fluorescein (Promega, Madison, WI, USA), to identify the cells with a high number of exposed 3� ends in their DNA [29], and counter stained with propidium iodide (1*g/ml in PBS �CaCl2, -MgCl2, Molecular Probes, Eugene OR, USA), which stains all cells in the tissue sections. The sections were finally treated with SlowFade�-Light Antifade (Molecular Probes, Eugene, OR, USA) cover medium. Next to the in vivo control experiments as listed above, positive and negative control sections were created during each staining session, by applying 100*l of DNase I (GibcoBRL, Burlington, ON, Canada) at random to one brain section prior to treatment with the TdT enzyme. DNase treatment of the fixed cells results in fragmentation of the chromosomal DNA and exposure of multiple 3� ends at which fluorescein-12-dUTP� is incorporated. The negative control section was created by incubation of a brain section with a buffer containing fluorescein-12-dUTP�, but no TdT enzyme.
Brain sections from each of the in vivo test groups were prepared in different staining sessions, to ensure that the observed results were not attributable to variations in the procedures during the different staining session.
A fluorescence confocal laser-scanning microscope (CLSM) (BioRad MRC 600, Mississauga, ON, Canada) providing 488 and 564nm excitation wavelength, was used for identification and quantification of the apoptotic cells. For presentation of apoptotic cells and bodies imaging software (Confocal Assistant Version 4.02, Copyright 1994-1996) was used to generate pseudo colour images, encoding propidium iodide- and Fluorescein-stained cells red and green, respectively.
Red blood cells take up the Fluorescein stain due to their lipophilic nature. Hence, the density of the apoptotic cells can not be calculate using imaging software, as the available software could not discern red blood cells from apoptotic cells on the basis of the green fluorescence intensity only. The slides were therefore scanned by eye enabling use of size and morphology additionally to fluorescence intensity for identification of apoptotic cell. Using a 200x magnification the CLSM provided a field of view of ~ 250x 250 mm. The entire brain section was scanned by moving the microscope stage in a millimetre grid pattern in x and y direction. Up to three sections from each brain were evaluated in this manner to ensure that the results were qualitative consistent. The number of apoptotic cell per field of view was entered into a 2D grid. Assuming that apoptotic cell density is a function of fluence rate, the geometrical centre of the apoptotic cell distribution was computed and subsequently the apoptotic cell density (#/mm2) and plotted on a semi-logarithmic scale versus radius (mm). Results of animals treated under similar conditions and of the different sections per animal were averaged prior to plotting the radial apoptotic cell density. The average radial fluence rate, as measured in the previous study [14] is also indicated in these apoptotic cell density plots to verify the assumption that apoptotic cell density is a function of fluence rate.
To confirm the incidence of apoptosis on the basis of cellular morphology, the tissue sections directly below each of those stained with Fluorescein were cut, stained with hematoxylin and eosin, and scanned under a light microscope for the incidence of cell shrinkage, karyhorrexis and pyknosis.
Results
The efficacy of the TdT assay was tested using the positive and negative control slides. Approximately 70% of the DNase-treated cells on the positive control slides demonstrated green fluorescence, verifying that the TdT assay was effective in labelling the exposed 3� ends of apoptotic DNA. Similarly, none of the negative control slides demonstrated green fluorescence, indicating that the incorporation of fluorescein-12-dUTP* was mediated by the TdT enzyme.
All control groups (I-III) showed comparable numbers of apoptotic cells per section. Groups I and II displayed an average of 3.7 apoptotic cells over the entire area of their brains. The contralateral hemispheres of each animal in Groups 1-8 demonstrated on average 2-3 apoptotic cells indicating that administration of either light or photosensitiser alone does not produce apoptosis 24 hours following PDT. This was confirmed by the respective H+E-stained sections showing neither necrosis nor cells with identifiably characteristic apoptotic morphologies.
Table 2 shows the total number of apoptotic cells present in the PDT-treated hemispheres for each animal and the average for similar treated animals. Using 514 nm as PDT-treatment wavelength, Photofrin produced the highest average apoptotic cell count of 52 +- for the entire brain section. ALA, mTHPC and SnET2 liposome produced intermediate effects with 15 +- , 15 +- and 15 +- average apoptotic cell count, respectively. This is equivalent to ~ 10000 apoptotic cells for Photofrin mediated PDT and 3000 apoptotic cells for the other three photosensitisers. AlClPc-mediated PDT resulted in apoptotic cell counts comparable with the control animals in the treated and contralateral hemisphere. The radius of necrosis in normal brain tissue was 2.6mm, 2.4mm, 3.1mm and 0.58 mm for animals of groups 2, 5, 6 and 8 respectively [14]. Two animals of Group 7 (SnET2) were treated at 660 nm and showed the highest apoptotic cell counts in the treated hemisphere. The same animals showed also the largest necrotic area, sometimes invading the contralateral side [14]. It should be noted here that 660nm light has an at least two times larger penetration depth in brain tissues and thus treats at least an eight times larger volume resulting in more swelling and oedema than light of 514nm wavelengths [13].
In most brain sections, the majority of the apoptotic cells were localised adjacent to the region of PDT-induced necrosis or in small pockets of viable cells within the necrotic tissue. Fig. 1 shows the radial distributions of apoptotic cells for animals in Groups 1, 2, 3, 4, and 6 based on the average for similar treated animals with up to 3 sections per animal. . For Photofrin and mTHPC mediated PDT the apoptotic cell density seems to follow the slope of the fluence rate as measured in a previous study [13] in a tumour bearing animal without photosensitizers. This trend is not as clear for ALA induced PpIX mediated PDT (figure 1b). Additionally, figure 1 shows the respective boundaries of necrosis as measured also previously [14]. The data suggests that apoptosis occurs, at a level above the control animals, also beyond the boundary of necrosis. No radial plots for groups 5, and 8 are shown due to the low number of observed apoptotic cells, and group 7 results are not shown due to the diffuse distribution of the apoptotic events, probably due to the massive damage and swelling observed in these animals following PDT.
The morphology of Fluorescein-stained cells is shown in Figure 2, which displays pyknotic chromatin packed in smooth masses against the cell membrane. Cell shrinkage, chromosomal fragmentation, as well as darkly-staining chromatin abutting along cellular nuclear envelopes is also shown in Figure 3 and 4 in H+E-stained sections, thus confirming the morphological observation in the fluorophore stained sections.
Discussion and Conclusion
Apoptotic cells and bodies are identified using the TdT assay, marking the exposed 3' ends of apoptotic DNA fragments produced by the activation of endogenous endonucleases [20], and histopathological study displaying the gross morphological characteristics of apoptotic cells. Apoptosis is never a completely synchronous process and cells at different stages coexist in the tissue sections [11]. Hence, the results of this study, using a single time point post PDT, will underestimate the total number of apoptotic cells present in any given brain section, as the in situ assay and the histopathology detect only the intermediate stages of apoptosis (DNA fragmentation, chromatin condensation, and formation of apoptotic bodies) but not the early or late phases of apoptosis (loss of membrane asymmetry [7], phagocytosis of apoptotic bodies by macrophages and neighbouring cells [17]). The methods used for detection do not allow for identification of the origin of an apoptotic body, but the total number of apoptotic cells is statistically not different between tumour bearing and normal animals (see figure 1). Thus, we assume that both normal brain cells and VX2 tumour cells undergo PDT-induced apoptosis.
The control animals treated with light or drug alone and the contralateral hemispheres exhibited on average less than 4 apoptotic cells in the entire area of the surveyed brain sections, indicating that light or photosensitiser alone do not induce apoptosis. These apoptotic cells probably occurred spontaneously as part of a homeostatic process that regulates cell proliferation and total cell number.
The spatial distribution of the apoptotic bodies ranges from close to a normal distribution to a circular distribution. Thus, the method of determining the geometric centre of apoptotic cell distribution may be somewhat arbitrary, but currently no other methods of determining the location of the source are available. Comparing the previously measured fluence rate distribution [14] with the computed radial apoptotic cell density suggests that, at least for Photofrin and mTHPC, fluence and/or fluence rate is a promoter of apoptosis. Conversely, no dependence between the total number of apoptotic cells or their radial distribution and the injected photosensitiser concentration was observed for Photofrin and ALA. Several studies have shown that Photofrin levels in brain tumours were at least 10 times that in normal brain [5, 14], but this difference in uptake is also not reflected in the apoptotic response to PDT. It is possible that at the low photosensitiser concentration all subcellular binding sites, capable of initiating apoptosis, are already filled for these two photosensitisers. The comparatively large apoptotic response associated with Photofrin may reflect the finding that the PDT threshold value of Photofrin is low, compared to the other photosensitizers tested, indicating high sensitivity of the brain tissue to PDT [14]. It was also shown in some cancer cell lines that apoptosis occurs as a direct result of Photofrin-mediated PDT in vitro [8].
With the exception of SnET2 (emulsion) treated with 660 nm light, no correlation between the radius of necrosis and number of apoptotic cells count was observed, suggesting that apoptosis does not result only following brain swelling and hypoxia [19], secondary effects of intracranial PDT.
The different results for PDT induced apoptosis for the different photosensitizers suggest that not only brain swelling, but also the cellular distribution of the photosensitiser under the here selected conditions. While the bulk photosensitiser uptake in tissue and fluence rate distribution may be adequate parameters to model tissue response due to PDT based on tissue necrosis, a model describing the spatial distribution of apoptosis clearly needs to consider next to these bulk parameters also microscopy, cellular and subcellular, distribution of the photosensitisers and their biochemical activation pathways. Lilge et al. [14] have shown that different brain structures express unique specific uptakes for different photosensitizers and several other studies have demonstrated that photosensitizers localise differently within cells [1, 2, 25]. Noodt et al. [20] noted during in vitro experiments that phthalocyanine derivatives may localise different from Photofrin and ALA-induced PpIX. Since singlet oxygen is short-lived and acts within less than 0.1�m of the site of its production [1], AlClPc could conceivably localise in places too remote from cellular sensors to activate the genetic program of apoptosis [20]. Another explanation, proposed by Ben-Hur et al. [2], is that phthalocyanine drugs may act through relative oxygen species other than singlet oxygen.
Recent studies on the effects of PDT with various photosensitizers on tumour vasculature showed that Photofrin-mediated PDT results in a drop of the vascular perfusion in murine tumours to less than 10% after 24 hours. In contrast, 24 hours following injection, mono sulfonated aluminium phthalocyanine (AlSPc) is selectively retained in tumour cells, [4, 21] and has little effect on tumour blood flow immediately after treatment and vascular perfusion is reduced by only 50% [4].
Thus, the apoptotic response produced 24 hours following Photofrin-mediated PDT may result from disruption of tumour vasculature and subsequent ischemic hypoxia, while the direct tumouricidal effects of phthalocyanine-mediated PDT may not result in apoptosis. It should be noted that Rabbits 2 and 3, treated with 1mg/kg SnET2 (emulsion), demonstrated the largest apoptotic response in the entire study and that this response was not localised in the region of necrosis. This response may not be a direct result of PDT, but reflects use of a treatment wavelength (660nm) with twice the penetrating depth, compared to 514 nm, hence capable of inducing necrosis and swelling in an eight times larger volume. Clinical use of PDT uses > 630nm light, equally capable to cause increased intracranial pressure due to brain inflammation and oedema possibly leading to the induction of a stronger apoptotic response.
Li et al. report that peak levels of apoptosis in the CNS following administration of ionising radiation to the rat spinal cord occur 8 hours after treatment and fall back to baseline levels after 24 hours [10]. While the results of the present investigation show that PDT-induced apoptosis in the brain have not returned to baseline levels at the 24-hour time point, it is unknown based on this study if PDT would elicit an overall greater apoptotic responses compared to ionising radiation. To evaluate also the biological significance, the origin of the apoptotic cells is important. Following ionising radiation, it has be shown that thymocytes exhibit a high rate of apoptosis, whereas oligodentrocytes show only a low incidence of apoptosis, so the latter are clinically of greater important.
Conclusion:
This study demonstrates that intracranial PDT performed on rabbits in vivo produces an apoptotic effect at 24 hours of treatment. The apoptotic effects produced by the five different photosensitizers suggest that apoptosis can extend beyond the volume of coagulative necrosis and that the specific number of apoptotic cells produced depend on the particular photosensitizer chosen, but not on photosensitizer dose, for the doses tested here. Hence photosensitizers like AlClPc and ALA may be preferable over Photofrin for intracranial PDT, when deep penetrating wavelengths of light are used or when normal brain structures are possibly be exposed.
The results of this retrospective preliminary analysis do not allow detailed analysis of the mechanism leading to apoptosis following intracranial PDT. PDT-induced apoptosis may occur as a result of either direct activation of an apoptotic pathway, or indirect due to damage to brain structures, ischemic hypoxia or brain swelling. Thus, this study resulted in more questions which stress the need for further research into the direct and indirect mechanism initiating apoptosis following PDT.
Acknowledgements:
The authors wish to thank Dr. Y.Q. Li for his help interpreting the H+E-stained slides, Poupak Pournazari, Anne Lee, Jane Park, and Marja Sepers for their assistance with the in vivo experiments, and Ralph DaCosta for help with the confocal microscope. Photofrin was kindly provided by Quadralogic Technologies (Vancouver, B.C., Canada), SnET2 (Pulytin) by Miravant Inc. (Santa Barbara, CA, USA), mTHPC by Scotia Drug Discovery (London, UK) and AlClPc by Dr. Nancy Oleinick (Case Western Reserve University, Cleveland, OH, USA). This work was supported in part by the National Institute of Health contract #POI-CA43692 and by Photonics Research Ontario.
Figure Legends:
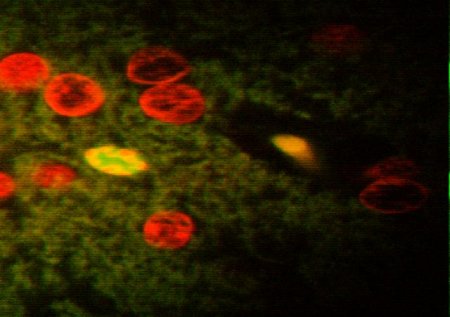 Figure 1(A-C): Semilog plot of apoptotic cell density (#/mm2) vs. radius (mm) for a) Photofrin, b) ALA-induced PpIX and mTHPC. Where applicable open symbols refer to the low drug dose and solid symbols to the high drug dose. Squares refer to results from tumour bearing animals, where triangles show the average of all animals treated with the respective condition. The vertical dashed and solid lines refers to the radii of necrosis for the low and high drug doses respectively, as computed in our previous study [14]. The thick sloped line is the fluence given in units of [mm mJ cm-2] as measured previously in vivo in a tumour bearing animal without photosensitizer.
Figure 1(A-C): Semilog plot of apoptotic cell density (#/mm2) vs. radius (mm) for a) Photofrin, b) ALA-induced PpIX and mTHPC. Where applicable open symbols refer to the low drug dose and solid symbols to the high drug dose. Squares refer to results from tumour bearing animals, where triangles show the average of all animals treated with the respective condition. The vertical dashed and solid lines refers to the radii of necrosis for the low and high drug doses respectively, as computed in our previous study [14]. The thick sloped line is the fluence given in units of [mm mJ cm-2] as measured previously in vivo in a tumour bearing animal without photosensitizer.
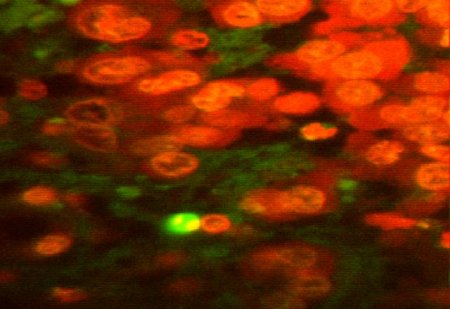 Figure 2:Confocal images of fluorescein-stained apoptotic cells (bright green) in brain sections of tumour-bearing animals (1000X mag.). The red cells are propidium iodide-stained VX2 tumour cells; a) animal administered 1mg/kg SnET2 in emulsion and treated with 50J of 660nm light; b) animal administered 2.5mg/kg Photofrin and treated with 50J of 514nm light.
Figure 2:Confocal images of fluorescein-stained apoptotic cells (bright green) in brain sections of tumour-bearing animals (1000X mag.). The red cells are propidium iodide-stained VX2 tumour cells; a) animal administered 1mg/kg SnET2 in emulsion and treated with 50J of 660nm light; b) animal administered 2.5mg/kg Photofrin and treated with 50J of 514nm light.
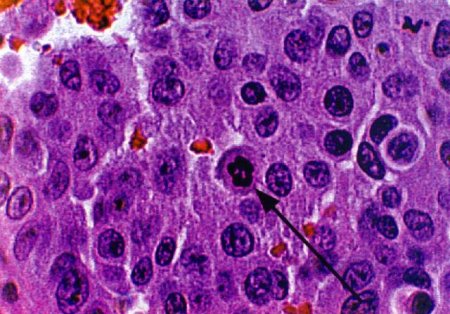 Figure 3:H+E-stained sections of tumour bearing brains treated with 50J of 514nm light (1000X mag.); a) animal administered 20mg/kg ALA and sacrificed 24 hours following PDT; b) animal administered 1mg/kg SnET2 (liposome) and sacrificed 72 hours following PDT, exhibiting condensed chromatin packed in smooth masses against the cell membrane (arrows).
Figure 3:H+E-stained sections of tumour bearing brains treated with 50J of 514nm light (1000X mag.); a) animal administered 20mg/kg ALA and sacrificed 24 hours following PDT; b) animal administered 1mg/kg SnET2 (liposome) and sacrificed 72 hours following PDT, exhibiting condensed chromatin packed in smooth masses against the cell membrane (arrows).
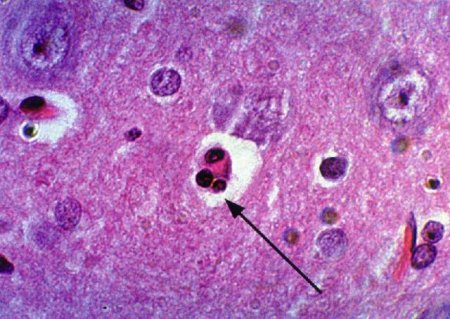 Figure 4:H+E-stained sections of tumour bearing brains treated with 50J of 514nm light (1000X mag.); a) animal administered 10mg/kg Photofrin and sacrificed 24 hours following PDT; b) animal administered 1mg/kg SnET2 (liposome) and sacrificed 72 hours following PDT, exhibiting sickle-shaped pyknotic chromatin, characteristic of apoptosis, packed into smooth masses against the nuclear envelope (arrows). Eosin-stained red blood cells are present with the regions of coagulative necrosis.
Figure 4:H+E-stained sections of tumour bearing brains treated with 50J of 514nm light (1000X mag.); a) animal administered 10mg/kg Photofrin and sacrificed 24 hours following PDT; b) animal administered 1mg/kg SnET2 (liposome) and sacrificed 72 hours following PDT, exhibiting sickle-shaped pyknotic chromatin, characteristic of apoptosis, packed into smooth masses against the nuclear envelope (arrows). Eosin-stained red blood cells are present with the regions of coagulative necrosis.
References
- BEN-HUR E, ROSENTHAL, I. (1985). The Pthalocyanines: a new class of mammalian cells photosensitizers with a potential for cancer phototherapy. Int. J. Radiat. Biol. 47(2): 145-147.
- BREMNER JCM, ADAMS GE, PEARSON JK, SANSOM JM, STRATFORD IJ, BEDWELL J, BOWN SG, MACROBERT AD, PHILLIPS D. (1992). Increasing the Effect of Photodynamic Therapy on the RIF-1 Murine Sarcoma, Using the Bioreductive Drugs RSU1069 and RB6145. Br. J. Cancer, 66: 1070-1076.
- CHAN WS, BRASSEUR N, LA MADELEINE C, VAN LIER JE. (1996). Evidence for Different Mechanisms of EMT-6 Tumor Necrosis by Photodynamic Therapy with Disulfonated Aluminum Phthalocyanine or Photofrin: Tumor Cell Survival and Blood Flow. Anticancer Research, 16: 1887-1892.
- CHOPP M, DERESKI MO, MADIGAN L, JIANG F, LOGIE B. (1996). Sensitivity of 9L Gliosarcomas to Photodynamic Therapy. Radiation Research, 146: 461-465.
- DERESKI MO, CHOPP M, GARCIA JH, HETZEL FW. (1991). Depth Measurements and Histopathological Characterization of Photodynamic Therapy Generated Normal Brain Necrosis as a Function of Incident Optical Energy Dose. Photochem Photobiol., 54: 109-112.
- van ENGELAND M, NIELAND LJ, RAMAEKERS FC, SCHUTTE B, REUTELINGSPERGER CP. (1998). Annexin V-affinity Assay: A Review on an Apoptosis Detection System Based on Phosphatidylserine Exposure. Cytometry, 31: 1-9.
- HE XY, SIKES RA, THOMSEN S, CHUNG LWK, JACQUES SL. (1994). Photodynamic Therapy with Photofrin II Induces Programmed Cell Death in Carcinoma Cell Lines. Photochem Photobiol., 59: 468-473.
- LAPERRIERE NJ, CEREZO L, MILOSEVIC MF, WONG CS, PATTERSON B, PANZARELLA T. (1997). Primary Lymphoma of Brain: Results of Management of a Modern Cohort with Radiation Therapy. Radiotherapy and Oncology, 43: 247-252.
- LI YQ, JAY V, WONG CS. (1996). Oligodendrocytes in the Adult Rat Spinal Cord Undergo Radiation-induced Apoptosis. Cancer Research, 56: 5417-5422.
- LI YQ, GUO YP, JAY V, STEWART PA, WONG CS. (1996). Time Course of Radiation-Induced Apoptosis in the Adult Rat Spinal Cord. Radiotherapy and Oncology, 39: 35-42.
- LI YQ, WONG CS. (1997). Radiation-induced Apoptosis in the Rat Spinal Cord: Lack of Equal Effect per Fraction. Int. J. Radiat. Biol, 71(4): 413-420.
- LILGE L, OLIVO MC, SCHATZ SW, MaGUIRE SA, PATTERSON MS, WILSON BC. (1996). The Sensitivity of Normal Brain and Intracranially Implanted VX2 Tumour to Interstitial Photodynamic Therapy. Br. J. Cancer, 73: 332-343
- LILGE L, WILSON BC. (1998). Photodynamic Therapy of Intracranial Tissues: A Preclinical Comparative Study of Four Different Photosensitizers. J. Clin. Laser Med. & Surg., 16(2): 81-91.
- MARGARON P, MADARNAS P, QUELLET R, VAN LIER JE. (1996). Biological Activities of Pthalocyanines. XVII Histopathologic Evidence for Different Mechanisms of EMT-6 Tumour Necrosis Induced by Photodynamic Therapy with Disulfonated Aluminum Pthalocyanine or Photofrin. Anticancer Research, 16: 613-620.
- MAHALEY MS, METTLIN C, NATARAJAN N, LAW ER, PEACE BB. (1989). National Survey of Patterns of Care for Brain-Tumor Patients. J. Neurosurg., 71: 826-836.
- MAJNO G, JORIS I. (1995). Apoptosis, Oncosis, and Necrosis: An Overview of Cell Death. Am. J. Path., 146: 3-15.
- McCaughan JS, ed. (1992) A Clinical Manual: Photodynamic Therapy of Malignancies, Chapter 13: Photodynamic Therapy for Brain Tumors. R.G Landes Co.: Boca Raton, FLA.
- MULLER P, WILSON BC. (1996). Photodynamic therapy for amlignant newly diagnosed supratentorial gliomas. J Clin. Laser. Surg. Med. 14:263-270
- NOODT BB, BERG K, STOKKE T, PENG Q, NESLAND JM. (1996). Apoptosis and Necrosis Induced with Light and 5-animolaevulinic Acid-Derived Protoporphyrin IX. Br. J of Cancer, 74: 22-29.
- PENG QM, NESLAND JM, REMINGTON C. (1990). Aluminum Pthalocyanines with Asymmetrical Lower Sulfonation and with Symmetrical Higher Sulfonation: a Comparison of Localizing and Photosensitizing Mechanism in Human Tumor LOX Xenografts. Int. J. Cancer, 46: 719-726.
- SCHULTHEISS TE, KUN LE, ANG KK, STEPHENS LC. (1995). Radiation Response of the Central Nervous System. Int. J. Radiation Oncology Biol. Phys., 31(5): 1093-1112.
- SHELINE GE, WARA WM, SMITH V. (1980). Therapeutic Irradiation and Brain Injury. Int. J. Radiation Oncology Biol. Phys., 6: 1215-1228.
- WHITE E. (1996). Pathway of Regulation of Apoptosis: Overview of Aptosis. Calbiochem- Novabiochem Int.: 8-15.
- WILSON BC, OLIVO M, SINGH G. (1997). Subcellular Localization of Photofrin and Aminolevulinic Acid and Photodynamic Cross-Resistance in Vitro in Radiation-Induced Fibrosarcoma Cells Sensitive or Resistant to Photofrin-Mediated Photodynamic Therapy. Photochem. Photobiol., 65(1).
- WONG CS, VAN DYK J, MILOSEVIC M, LAPERRIERE NJ. (1994). Radiation Myelopathy Following Single Courses of Radiotherapy and Retreatment. Int. J. Radiation Oncology Biol. Phys., 30(3): 575-581.
- YOSHIDA Y, DERESKI MO, GARCIA JH, HETZEL FW, CHOPP M. (1992). Neuronal Injury After Photoactivation of Photofrin II. Am. J of Path., 141(4): 989-997.
- PATTERSON MS, WILSON BC, GRAFF R (1990) In vivo test of the concept of photodynamic threshold dose in normal rat liver photosensitized by aluminium chlorosulphonated phthalocyanine. Photochem Photobiol. 51:343-349
- LI X, TRAGANOS F, MELAMED MR, DARZYNIKIEWICZ Z (1995) Single-step procedure for labeling DNA stand breaks with fluorescein- or BODIPY-conjungated deoxynucleotides: detection of apoptosis and bromodeoxyuridine incorporation. Cytometry 20:172-180 (1995)
| Discussion Board | Previous Page | Your Symposium |