Invited Symposium: Hypertension III: Flow-Induced Vascular Remodeling
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
It is intuitively clear that elevated pressure could give rise to hypertrophy through an increase in wall tension, but protection from elevated pressure also prevents inward, eutrophic remodeling of the arterioles. To examine the mechanism behind these changes, we studied isolated, small arteries exposed to elevated pressure[4]. As shown in figure 1, raising the pressure in an isolated first order mesenteric artery to 140 mmHg induced the expression of protooncogenes and 18S ribosomal RNA.
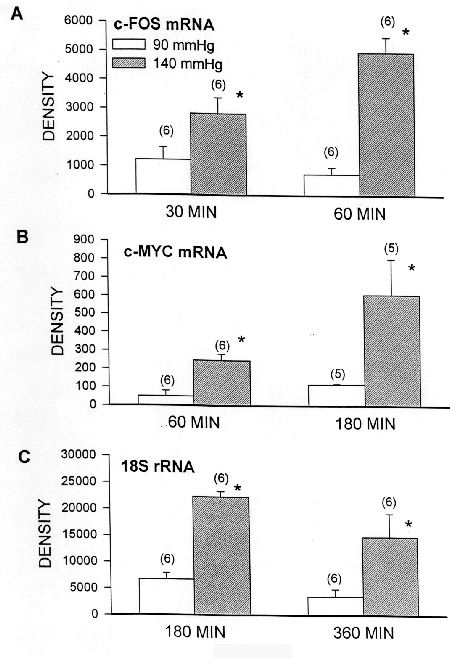 Fig. 1:Expression of c-fos and c-myc mRNA and 18S rRNA in isolated mesenteric arteries maintained at the times indicated at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
Fig. 1:Expression of c-fos and c-myc mRNA and 18S rRNA in isolated mesenteric arteries maintained at the times indicated at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
Beta actin did not differ from controls maintained at 90 mmHg (not shown). When Allen et al.[5] used slightly smaller mesenteric arteries in a second study, some were capable of a myogenic response that reduced wall stress. By raising luminal pressure to 165 mmHg a range of wall stresses were obtained and it was found that expression of c-myc and 18S rRNA correlated with wall stress (Figures 2 and 3) but not with the diameter expressed as a percentage of the relaxed diameter. This latter designation is an index of cell stretch as any vessel with a diameter less than 100% of the relaxed diameter has tone due to smooth muscle contraction.
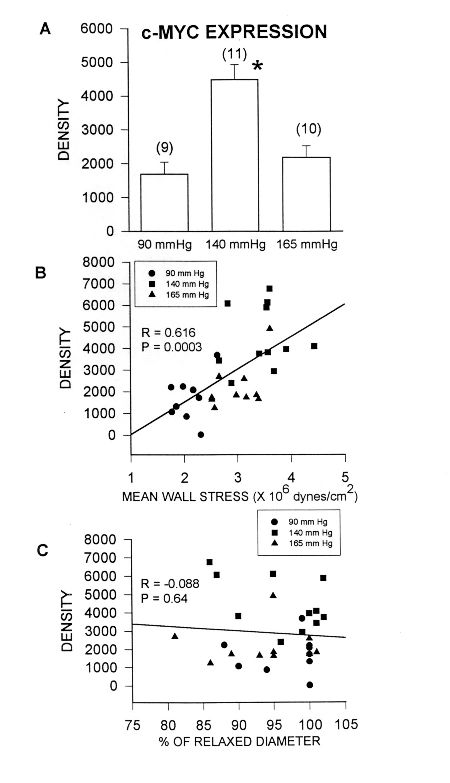 Fig. 2:Expression c-myc mRNA in isolated mesenteric arteries maintained for 3 hours at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
Fig. 2:Expression c-myc mRNA in isolated mesenteric arteries maintained for 3 hours at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
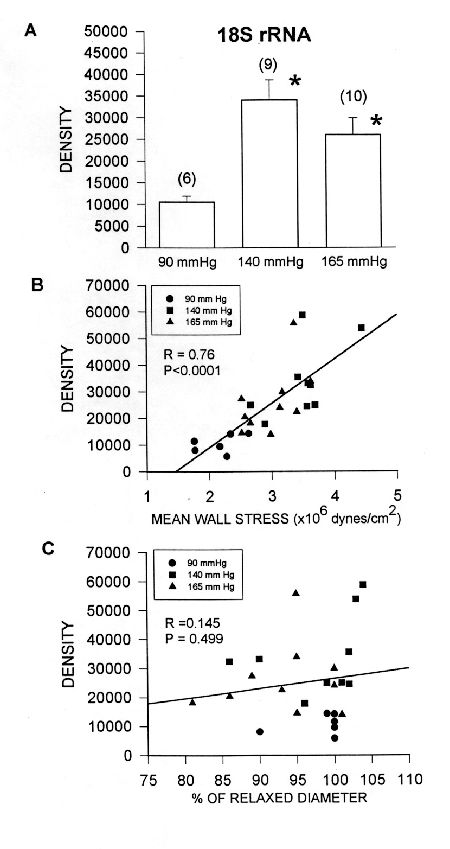 Fig. 3:Expression of 18S rRNA in isolated mesenteric arteries maintained for 3 hours at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
Fig. 3:Expression of 18S rRNA in isolated mesenteric arteries maintained for 3 hours at 90 mmHg or 140 mmHg intraluminal pressure. Gene expression was measured by in situ hybridization quantitated on a phosphorimager in arbitrary density units.
The implications of these results for remodeling of arteries in hypertension is that the severe vasoconstriction taking place in the arterioles during the onset of hypertension may ameliorate the increase in wall stress and prevent the development of hypertrophy. Instead, the resistance arteries and arterioles undergo inward, eutrophic remodeling. This series of events was demonstrated by Hashimoto et al.[6] who showed that tone was very high in the cremasteric arterioles of one-kidney, one-clip hypertensive rats (1K1C) during the first 2 weeks of hypertension. The tone decreased toward control levels by 8 weeks, but the lumen of the arterioles was structurally reduced, thus maintaining increased vascular resistance without the necessity of increased active tone.
If the lumen size of an artery is regulated by flow rate through shear stress, why is the lumen of these vessels reduced when it is well known that the cardiac output and its distribution to skeletal muscle is normal? Wang et al.[7] measured flow and shear rate in the first-order arteriole of the cremaster muscle in the 1K1C rat and found that at 1 week of hypertension the shear rate was not elevated because the blood flow was reduced (Figure 4). By 4 weeks, however, the flow rates were back to normal and the wall shear rate was significantly increased. Assuming the viscosity was the same as in controls, this means that the shear stress on the endothelial cells was elevated, thus unbalancing the relationship between shear stress and lumen size in this arteriole.
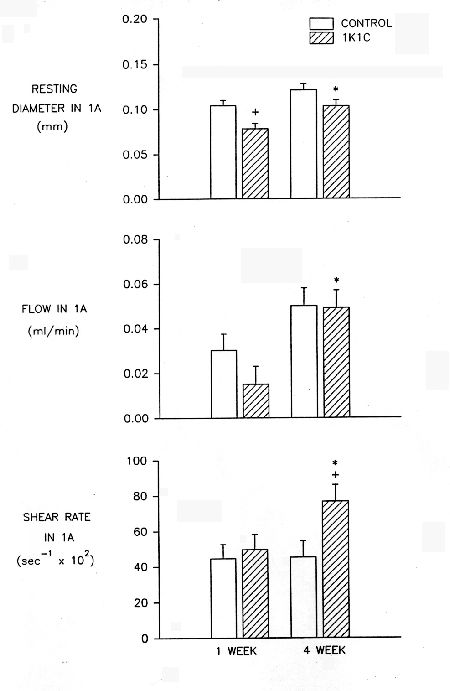 Fig. 4:Resting diameter, blood flow and wall shear rate in first-order arterioles of control and 1K1C hypertensive rat. Red cell velocity was measure by the dual-slit technique and blood flow and shear rate were calculated from velocity and diameter data.
Fig. 4:Resting diameter, blood flow and wall shear rate in first-order arterioles of control and 1K1C hypertensive rat. Red cell velocity was measure by the dual-slit technique and blood flow and shear rate were calculated from velocity and diameter data.
Endothelial dysfunction in hypertension is now well known. Nakamura et al.[8] showed in this same 1K1C model of 4 weeks duration that the response to the endothelial dependent vasodilator, acetylcholine, was impaired (Figure 5).
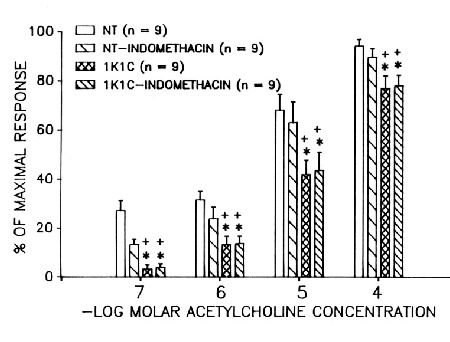 Fig. 5:Vasodilation of arcade arterioles of the spinotrapezius muscle of control and 1K1C hypertensive rats in response to increasing doses of acetylcholine. Vasodilation is expressed as the percentage of that obtained with 3 mM adenosine. Indomethacin was in the superfusion solution at a concentration of 2.8 mM.
Fig. 5:Vasodilation of arcade arterioles of the spinotrapezius muscle of control and 1K1C hypertensive rats in response to increasing doses of acetylcholine. Vasodilation is expressed as the percentage of that obtained with 3 mM adenosine. Indomethacin was in the superfusion solution at a concentration of 2.8 mM.
Pretreatment with indomethacin decreased the vasodilatory response in controls but not in 1K1C arteries, indicating that the release of nitric oxide is probably reduced. The response to bradykinin was also reduced in the 1K1C arterioles (Figure 6), and the response was reduced in both controls and hypertensives by indomethacin.
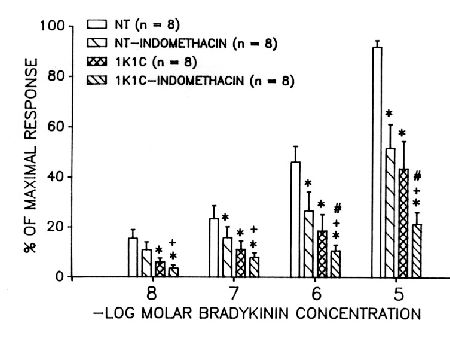 Fig. 6: Vasodilation of arcade arterioles of the spinotrapezius muscle of control and 1K1C hypertensive rats in response to increasing doses of bradykinin. Vasodilation is expressed as the percentage of that obtained with 3 mM adenosine. Indomethacin was in the superfusion solution at a concentration of 2.8 mM.
Fig. 6: Vasodilation of arcade arterioles of the spinotrapezius muscle of control and 1K1C hypertensive rats in response to increasing doses of bradykinin. Vasodilation is expressed as the percentage of that obtained with 3 mM adenosine. Indomethacin was in the superfusion solution at a concentration of 2.8 mM.
Thus the bradykinin-stimulated release of prostaglandins by the endothelial cells was essentially normal. The response to sodium nitroprusside did not differ between control and 1K1C arterioles, indicating that it is not the response by the vascular smooth muscle cells that is impaired, but the release of nitric oxide by the endothelial cells. Impaired release of nitric oxide by dysfunctional arteriolar endothelial cells in the 1K1C rats can explain why the shear stress is elevated and fails to stimulate the vessel to dilate and grow larger.
In summary, arterial remodeling in hypertension is the result of a combination of additive and opposing factors. Elevated pressure acting through wall stress promotes hypertrophy of the vascular wall, the characteristic response of large, non-myogenic arteries. Myogenic, neurogenic and autoregulatory tone in arterioles opposes the increase in wall stress during the development of hypertension. The reduction in lumen size is first maintained by the elevated tone, but over time the chronic reduction in diameter becomes a structural change. As the increasing arterial pressure forces more blood flow through the constricted artery, the dysfunction of the endothelial cells prevents the shear stress from stimulating luminal expansion. This interplay of mechanical forces also takes place in the normal vasculature to bring about remodeling during growth of the individual, uterine growth in pregnancy and chronic changes in physical activity. The arterial remodeling during the development of hypertension is not pathological but merely an adaptation to the mechanical forces imposed on the vessels in the face of their main goal to autoregulate blood flow.
References
- Folkow, B., M. Gurevich, M. Hallback, Y. Lundgren and L. Weiss, (1971) The hemodynamic consequences of regional hypotension in spontaneously hypertensive and normotensive rats. Acta Physiol Scand, . 83(4): 532-41.
- Bund, S. J.,K. P. West and A. M. Heagerty, (1991) Effects of protection from pressure on resistance artery morphology and reactivity in spontaneously hypertensive and Wistar-Kyoto rats Circ Res, . 68(5): 1230-40.
- Stacy, D. L. and R. L. Prewitt, (1989) Attenuated microvascular alterations in coarctation hypertension. Am J Physiol, . 256(1 Pt 2): H213-21.
- Allen, S. P., H. M. Liang, M. A. Hill and R. L. Prewitt, (1996) Elevated pressure stimulates protooncogene expression in isolated mesenteric arteries. Am J Physiol, . 271(4 Pt 2): H1517-23.
- Allen, S. P., S. S. Wade and R. L. Prewitt, (1997) Myogenic tone attenuates pressure-induced gene expression in isolated small arteries. Hypertension, . 30(2 Pt 1): 203-8.
- Hashimoto, H., R. L. Prewitt and C. W. Efaw, (1987) Alterations in the microvasculature of one-kidney, one-clip hypertensive rats. Am J Physiol, . 253(4 Pt 2): H933-40.
- Wang, D. H., R. L. Prewitt and C. K. Reilly, (1993) Altered local regulation of blood flow and shear rate in renal hypertension. Am J Hypertens, . 6(10): 851-6.
- Nakamura, T. and R. L. Prewitt, (1992) Alteration of endothelial function in arterioles of renal hypertensive rats at two levels of vascular tone. J Hypertens, . 10(7): 621-7.
| Discussion Board | Previous Page | Your Symposium |