Pharmacology & Toxicology Poster Session
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
The World Health Organization estimates that nearly 500,000 people in sub-Saharan Africa are infected with trypanosomiasis, also known as sleeping sickness [1]. At present the disease is resurging with prevalence levels reminiscent of the great epidemic of the 1930s, which killed half a million people. If untreated African sleeping sickness is always fatal. Available drug therapy is unsatisfactory because of the need for parenteral administration , the emergence of resistant parasites, and severe toxicity - between 5 and 10 % of the patients die as a direct result of melarsoprol administration [2]. Clearly, better drugs are urgently needed.
Sleeping sickness is caused byĀTrypanosoma brucei, known as African trypanosomes. After a bite by the tse-tse fly the parasites enter the bloodstream of the human host, multiply, and eventually invade the brain. Remarkably, they have an extremely reduced metabolism in which glycolysis to the stage of pyruvate is their only source of energy. In vitro a combination of glycerol and SHAM, which blocks glycolysis indirectly, kills the parasites within minutes [3]. However, no selective inhibitors that block trypanosomal glycolytic enzymes while leaving the equivalent human enzymes unaffected are available. Therefore, we decided to develop trypanosomatid-selective inhibitors by structure-based drug design methods.
Mathematical models show that in the bloodstream form of trypanosomes glyceraldehyde-3-phosphate exerts considerable control on the glycolytic flux, and appears therefore to be a good drug design target [4]. The crystal structures of both the parasite and the human enzyme have been solved [5,6]. While their structures are identical in the active sites, distinct structural differences occur in and around the binding pocket for the adenosine moiety of the NAD cofactor. NAD exhibits a weak affinity for both the parasite and human enzyme, with Km values of 0.45 and 0.04 mM, respectively. Hence, it is no surprise that adenosine is an even weaker competitive inhibitor, with a Ki of 50 mM [7]. Despite the widespread prejudice against millimolar leads we picked adenosine as a starting point for developing selective inhibitors that would outcompete the cofactor of trypanosomal GAPDH.
Unfortunately, T. brucei GAPDH can only be obtained by a laborious procedure that involves infecting rats; overexpression attempts were unsuccessfu l, a major drawback for structure-based drug design. Because GAPDH of Leishmania mexicana, a parasite related to T. brucei, does not exhibit this problem we used it as a substitute in many stages of our studies. This proved to be a good strategy as our subsequent structure determination of the L. mexicana enzyme showed that their adenosine pockets are virtually identical, with a 0.2 A rms deviation on backbone and 0.5 A on side chain atom positions [8].
Materials and Methods
COMPUTER MODELING
The three-dimensional structures of potential inhibitors were constructed interactively in the T. brucei GAPDH structure using the molecular modeling program BIOGRAF [9]. Subsequently, the most promising inhibitors were docked by Monte Carlo methods with the QXP software [10].
CRYSTALLOGRAPHY
Co-crystals of L. mexicana GAPDH with N6-benzyl-NAD were grown in sitting drops. Cryo-data to 3.4 A resolution were collected at SSRL beam line 7-1 and the structure was solved by molecular replacement methods. Rigid-body refinement of the four enzyme subunits lowered the R-factor to 30%. Because of the limited resolution, individual atomic positional refinement was not pursued. Fourfold non-crystallographic symmetry averaging of only the protein improved the electron density map considerably and resulted in unambiguous density for the modified cofactor.
ENZYME ASSAYS
GAPDH activity was measured in the direction of NADH formation by monitoring absorption at 340 nm. The reaction mixture contained 0.8 mM GAPDH and 0.19 mM NAD. All designed compounds were tested against L. mexicana GAPDH. The best inhibitors were assayed against T. brucei GAPDH.
BIOLOGICAL ASSAYS
Strain 427 T. brucei was obtained from K. Stuart (SBRI, Seattle, WA) and the bloodstream form of the parasites was cultured in HMI-9 medium containing 10% fetal calf serum. Murine 3T3 fibroblast cells were grown in monolayers. The growth of parasite and human cells was determined with Alamar Blue (TM). Alamar Blue quantitation of T.brucei was verified to correspond with visual counts determined with a hemacytometer.
Excretion of pyruvate from bloodstream form T. brucei into the media was determined in cells from a mid-log culture. Pyruvate was quantified by conversion into lactate with lactate dehydrogenase.
Results
SELECTIVITY
The NAD adenosine binding environment differs substantially between T. brucei and human GAPDH. In the parasite enzyme there exists a hydrophobic cleft adjacent to the ribose O2'. This cleft is absent in the human enzyme due to a different protein backbone conformation. Hence, this cleft provides an excellent opportunity to convey selectivity to our designed inhibitors. Docking studies with various hydrophobic ring systems quickly showed that only a benzene ring is narrow enough to fit in the cleft. However, it was not immediately clear how such a ring might be linked to the adenosine ribose. A link via O2' appeared inappropriate as this hydroxyl is a hydrogen bond donor to Asp 37. Thus, creating an ether or ester linkage would come with a high desolvation price. Modeling of various N2' derivatives revealed that an amide linker would be a proper hydrogen bond donor substitute and orient the benzene ring into the cleft. Also, it appeared that extra hydrophobic interactions could be picked up by introducing a methoxy substituent of the aromatic ring. Figure 1 shows how the designed inhibitor 2'-deoxy-2'-(3-methoxybenzamido)-adenosine fits beautifully into the selectivity cleft of T. brucei GAPDH.
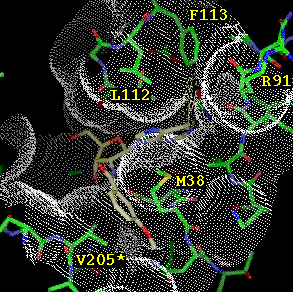 Figure
1: Binding mode model of 2'-deoxy-2'-(3-methoxybenzamido)adenosine
to T. brucei GAPDH, with the solvent accessible surface shown as
dots. The 3-methoxybenzamido moiety fits in the selectivity cleft formed
by Met 38 and Val 205*.
Figure
1: Binding mode model of 2'-deoxy-2'-(3-methoxybenzamido)adenosine
to T. brucei GAPDH, with the solvent accessible surface shown as
dots. The 3-methoxybenzamido moiety fits in the selectivity cleft formed
by Met 38 and Val 205*.
Enzyme inhibition studies showed that 2'-deoxy-2'-(3-methoxybenzamido)adenosi ne inhibits T. brucei GAPDH 45 times better than adenosine. More importantly , no evidence of human GAPDH blocking could be detected. Thus, we succeeded in obtaining a selective inhibitor [7].
AFFINITY BY SCREENING
Having solved the selectivity problem, we then focused on improving the affinity of our lead [11]. Modeling showed that the introduction of a 2-thienyl at position 8 of the purine ring would bury a major part of the Leu 112 side chain (Figure 1). After synthesis we discovered that a 180 fold gain in affinity with respect to adenosine was obtained this way. Unfortunately, 8-adenine substituents appeared in computro to be sterically incompatible with 2'-(3-methoxybenzamido) because they clash with the amido oxygen. Experimental evidence for this conclusion came after we subsequently synthesized 2'-deoxy-2'-(3-methoxybenzamido)-8-(2-thienyl)adenosine. This compound was only a millimolar inhibitor, worse than its mono-substituted parent compounds.
Subsequently, we shifted our attention to the N6 position of the purine. This atom is adjacent to two hydrophobic areas on the protein surface, one formed by the side chains of Leu 112, Phe 113 and Arg 91, another one by Met 38 and Arg 91 (Figure 1). Therefore, we purchased five N6-adenosine derivatives with hydrophobic substituents for screening, and synthesized nine others. Incorporating N6 into an amide function appeared to be detrimental, but several amines worked fine. Especially N6-benzyl-adenosine looked promising with a 10-fold affinity gain over adenosine.
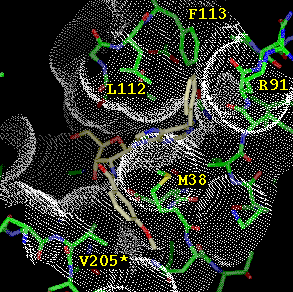 Figure
2: Modeled binding mode of N6-benzyl-adenosine to T. brucei
GAPDH, with the solvent accessible surface shown as dots. The N6-benzyl
group fits in a hydrophobic environment. Note that an alternative binding
model is possible in which the benzyl fits in between the Met 38 and Arg
91 side chains.
Figure
2: Modeled binding mode of N6-benzyl-adenosine to T. brucei
GAPDH, with the solvent accessible surface shown as dots. The N6-benzyl
group fits in a hydrophobic environment. Note that an alternative binding
model is possible in which the benzyl fits in between the Met 38 and Arg
91 side chains.
The benzyl substituent of N6-benzyl-adenosine can be modeled into the binding site in two different orientations (Figure 2). We resolved this dilemma by a crystal structure determination. Because we co-crystals of parasite GAPDH with N6-benzyl-adenosine failed to grow, we synthesized the N6-benzyl-NADĀanalogue and obtained crystals. In the experimental structure with L. mexicana GAPDH the N6-benzyl is sandwiched between the side chains of Met 39 and Arg 92, corresponding to Met 38 and Arg 91 in T.brucei (Figure 3).
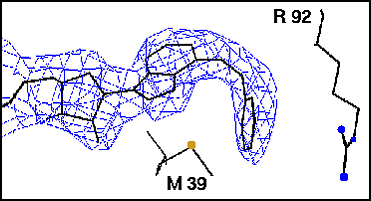 Figure
3: Experimental binding mode of N6-benzyl-NAD to L.mexicana
GAPDH.
Figure
3: Experimental binding mode of N6-benzyl-NAD to L.mexicana
GAPDH.
AFFINITY BY DESIGN
N6-Benzyl-adenosine formed the basis for further affinity improvement by design. A search in the Available Chemicals Database 95.2 revealed the commercial availability of 1,124 benzylamines of which 88 appeared suitable for reaction with 6-chloropurine riboside. All 88 were modeled as N6-adenosine derivatives in the T. brucei binding site. Poorly fitting molecules were rejected. After similarity clustering we decided on the synthesis of six of them. All six proved to be more potent than N6-benzyl-adenosine (Table 1). The best compound was N6-(1-naphtalenemethyl)adenosine, with a 333-fold gain in affinity over adenosine.
Table 1: Inhibition of L.mexicana GAPDH by N6-adenosine derivatives
N6-substituent IC50(ĄM) benzyl 4,2002-methylbenzyl 700
3-methylbenzyl 750
1,2,3,4-tetraH-1-naphtyl 360
1-naphtalenemethyl 150
2-[2-(hydroxymethyl)phenylthio]-benzyl 340
diphenylmethyl 240
COMBINING SELECTIVITY WITH AFFINITY
Several of the N6-substituents were combined with the 2'-(3-methoxybenzamido) substituent, leading to potent and selective inhibition (Table 2).
Table 2: Selective parasite GAPDH inhibition by N6-substituted
2'-deoxy-2'-(3-methoxybenzamido)adenosine derivatives (IC50 in ĄM)
N6-substituent L. mexicana T. brucei humana insoluble above and non-inhibitory at stated concentrationbenzyl 16 159 >530a2-methylbenzyl 4 40 >270a
1-naphtalenemethyl 0.2 2 >200a
In particular the 1-naphtalenemethyl derivative affords submicromolar inhibition. Its modeled binding mode is shown in Figure 4. None of the designed compounds inhibited the human GAPDH to any degree at submillimolar concentrations, their upper solubility limit.
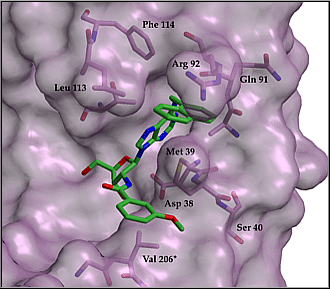 Figure
4: Modeled binding mode of 2'-deoxy-2'-(3-methoxybenzamido)-N6- (1-naphtalen
e-methyl)adenosine
to L.mexicana GAPDH.
Figure
4: Modeled binding mode of 2'-deoxy-2'-(3-methoxybenzamido)-N6- (1-naphtalen
e-methyl)adenosine
to L.mexicana GAPDH.
BIOLOGICAL ACTIVITY
Several of our designed compounds were tested for their potency in inhibiting parasite growth. Encouragingly, the ranking of the ED50 values thus obtained and the IC50 values in the enzyme inhibition assay was identical. The most potent GAPDHĀinhibitor, produced 50% growth inhibition of bloodstream form T. brucei at a concentration of 30 ĄM. Simultaneously, pyruvate excretion by the parasite was shut down in a dose-dependent manner. Importantly, none of our designed GAPDH inhibitors was toxic for the mammalian cells we assayed at concentrations up to 0.05 mM.
Discussion and Conclusion
The present study underlines the value of a structure-based approach to dramatically improve the affinity of a 50 millimolar lead. Such leads are generally ignored by the pharmaceutical industry, which insists on micromolar leads. The fact that we obtained five orders of magnitude affinity gain in one round of drug design shows that more optimism may be warranted. Also, the current results demonstrate that selectivity can be built into inhibitors.
During the course of this project many collegues have expressed concerns of using adenosine as a scaffold because of nature's ubiquitous use of this moiety in cofactors and co-substrates, such as NAD, FAD, ATP, etc. However, our 200 nM inhibitor's shape is a very significant departure from that of unmodified adenosine. In fact, it may be considered structurally as different from adenosine as an HIV-protease inhibitor from poly-alanine. Furthermore, we have tested our best inhibitor against several ATP- and NAD-dependent enzymes, such as phosphoglycerate kinase, lactate dehydrogenase and glycerol-3-phosphate dehydrogenase, and found no inhibition.
Our optimism is further corroborated by the first in vitro test results. The most potent GAPDH inhibitors were most toxic for the parasites and blocked pyruvate production. None of these effects were seen in the parallel studies with mammalian cells. While the current results are highly suggestive we cannot rule out the possibility that our inhibitors kill the parasites by other mechanisms. Studies to resolve this issue are underway.
ACKNOWLEDGEMENTS
Thanks go to the following collaborators at the University of Washington: Alex Aronov and Dr. Michael Gelb for synthesis of the inhibitors and enzymatic assays, Dr. Stephen Suresh for the crystal structure determination of the complex between N6-benzyl-NAD and L. mexicana GAPDH, Dr. Frederick Buckner for assessing the potency of the inhibitors in vitro, and Dr. Wim Hol for leading the project. This project has also benefited in earlier stages from contributions by Dr. Paul Michels, Dr. Veronique Hannaert and Dr. Fred Opperdoes (Christian de Duve Institute of Cellular Pathology, Brussels, Belgium), Dr. Serge Van Calenbergh, Dr. Arthur Van Aerschot and Dr. Piet Herdewijn (REGA Institute, Leuven, Belgium).
References
- Smith DH, Pepin J, Stich AHR (1998). Human African trypanosomiasis: an emerging public health crisis. Brit. Med. Bull. 54:341-355.
- Pepin J, Milord F (1994). The treatment of human African trypanosomiasis. Adv. Parasitol. 33:1-47.
- Clarkson AB, Brohn FH (1976). Trypanosomiasis: an approach to chemotherapy by the inhibition of carbohydrate metabolism. Science 194:204-206.
- Bakker BM (1998). Control and regulation of glycolysis in Trypanosoma brucei. Ph.D. Thesis, Free University of Amsterdam, The Netherlands.
- Vellieux FMD, Hajdu J, Verlinde CLMJ, Groendijk H, Read RJ, Greenhough TJ, Campbell JW, Kalk KH, Littlechild JA, Watson HC, Hol WGJ (1993). Structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei determined from Laue data. Proc. Natl. Acad. Sci. USA 90:2355-2359.
- Mercer WD, Winn SI, Watson HC (1976). Twinning in crystals of human skeletal muscle D-glyceraldehyde-3phosphate dehydrogenase. J. Mol. Biol. 104:277-283.
- Verlinde CLMJ, Callens M, Van Calenbergh S, Van Aerschot A, Herdewijn P, Hannaert V, Michels PAM, Opperdoes FR, Hol WGJ (1994). Selective inhibition of trypanosomal glyceraldehyde-3-phosphate dehydrogenase by protein structure-based design: Towards new drugs for the treatment of sleeping sickness. J. Med. Chem. 37:3605-3613.
- Kim H, Feil IK, Verlinde CLMJ, Petra PH, Hol WGJ (1995). Crystal structure of glycosomal glyceraldehyde-3-phosphyate dehydrogenase from Leishmania mexicana: Implications for structure-based drug design and a new position for the inorganic phosphate binding site. Biochem. 34:14975-14986.
- BIOGRAF. Molecular Simulations, Inc., San Diego, CA, USA.
- McMartin C, Bohacek R (1997). QXP: Powerful, rapid computer algorithms for structure-based drug design. J. Comput.-Aided Mol. Des. 11:333-344.
- Aronov AM, Verlinde CLMJ, Hol WGJ, Gelb MH (1998). Selective new inhibitors of trypanosomal glyceraldehyde-3-phosphate dehydrogenase via structure-based drug design. J. Med. Chem. 41: 4790-4799.
| Discussion Board | Previous Page | Your Poster Session |