Invited Symposium: Signal Transduction in Endothelium: Mechano-Sensing, Ion Channels and Intracellular Calcium
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Calcium: A Multifunctional Messenger In Endothelium
Communication between cells is one of the most important properties within the organism which is initiated by a variety of signals received at the cell surface. These signals affect via specific second messengers a plethora of cell functions such as secretion, motility, cell division, gene expression, and cellular metabolism. Among such second messengers, cytosolic Ca2+ is one of the most important signals, which is very well conserved throughout the phylogentic tree.
Endothelial cells (ECs) form an ideal anticoagulative surface for blood flow but importantly act as a multifunctional, signal-transducing surface. ECs regulate prevent blood clotting but can also trigger it in response to various signals, and can exert thrombolytic as well as thrombogenic activity. As antigen-presenting cells, they are also involved in immune responses. By changing the contractile state and by modulation of cell-cell contacts, they control the permeability of the blood-tissue interface. ECs initiate angiogenesis, vessel repair and adjust the vessel diameter to the hemodynamic needs. These multiple functions are mediated by the production and release of a variety of vasoactive agents, which affect the cells in the vessel wall or its immediate vicinity, including the endothelial cells themselves. Most importantly, among these substances include nitric oxide is NO or endothelium-derived relaxing factor (EDRF). Other compounds are endothelium-derived hyperpolarizing factor (EDHF), various pro staglandins (most importantly prostacyclin, PGI2), endothelins (ET), natriuretic peptide, small signaling molecules such as substance P, platelet activating factor (PAF), ATP, growth factors, steroids, and even larger proteins. Such proteins comprise factors involved in the blood clotting cascade, such as tPA (tissue plasminogen activator), TFPI (tissue factor pathway inhibitor), Von Willebrand factor (for a review, see 17, 28, 29, 30, 41). ECs respond not only to humoral substances, which bind to receptors, but also to mechanical forces, due to changes in flow rate (shear stress) or blood pressure (biaxial tensile stress) (6,7,8,23).
For all these -often functionally opposite responses - an intracellular increase of the Ca2+ concentration is the most decisive trigger. However, the regulation of these important Ca2+ signals and their connection to secretory events in ECs are not known. Two types of ca signals can be phenotypically differentiated: 1. A biphasic response of [Ca2+]i consisting of a fast peak and a long lasting plateau (18, 19, 41, 42), 2. Oscillations of [Ca2+]i and Ca2+-waves (19, 3, 26, 41). Both types of Ca2+ -signals seem to depend on different mechanisms. For the biphasic response, the release of Ca2+ from intracellular stores mainly reflects the fast peak in the biphasic Ca2+-response and activation of Ca2+ influx occurring from the extracellular space is responsible for the plateau. For the latter process ion channels are activated which provide functionally different influx routes for Ca2+ (28, 30, 41) and regulate and fine-tune the inwardly driving force for this Ca2+-influx (41). In this review only mechanisms will be discussed which contribute to the sustained, long-lasting plateau after agonist stimulation of ECs.
Mechanically Activated Ca2+ -Entry
One of the most fascinating properties of endothelial cells (ECs) is their ability to function as mechano-sensors. They are constantly exposed to changes of mechanical forces within a blood vessel and can respond to such forces. The best-studied fast response to mechanical stimulation is a release of nitric oxide, NO, which is preceded by a Ca2+ transient. Other fast responses comprise channel activation, synthesis of second messengers, activation of G-proteins. A direct activation of Ca2+-entry by shear stress has been reported from our group (47).
We have tried to analyse some of the mechano-sensor elements in ECs. Figure 1 shows a typical group of results. If an endothelial cell is mechanically stimulated in a Ca2+ free solution, it responds with activation of a Ca2+ transient (figure 1A). Because of the absence of extracellular Ca2+, it must be related to Ca2+ release. This release can be activated by all kinds of mechano-stimulation: shear stress, cell swelling, and cell stretch (43). If after mechano-stimulation an agonist is applied, no further Ca2+ signal can be activated, pre-treatment of the cells with thapsigargin also attenuates any further release of Ca2+. This indicates that a) the mechanical stimulus releases Ca2+ from Ins(1,4,5)P3 sensitive stores and b) this stimulation supposedly depletes these stores. This store depletion probably activates Ca2+-entry channels (store operated Ca2+-current, SOC, or Ca2+-release activated -calcium current, CRAC). We have described that this release of Ca2+ might be mediated via production of arachidonic acid (AA) because a) PLC not involved, b) heparin does not block this release , c) PLA2 blockers inhibit this release, and d) AA application mimics the effects of mechano-stimulation even in the presence of heparin and PLA2 blockers. The PKA inhibitory peptide is inefficient, as well as cyclooxygenase and lipoxygenase inhibition (for details see 43).
Mechanical stimulation of EC probably depletes the Ins(1,4,5)P3 sensitive Ca2+ stores. As a consequence of this depletion, Ca2+-entry is activated. Currents, which are activated by this mechanism, will be described in detail later. Experimental evidence for activation of Ca2+-entry due to mechano-stimulation is depicted in Figure 1B. Mechano-stimulation induces a Ca2+-transient as already shown in figure 1A but also activates a current which is independent on the changes in [Ca2+]i. This current is identical with the volume-regulated, Ca2+-independent anion (Cl- ) current (VRAC) and has been described in detail elsewhere (32, 33, 34, 43). VRAC is obviously a mechano-sensitive channel. After switching to a Ca2+ free solution and reapplication of Ca2+ (10 mM) a large Ca2+-signal appeared which reflect Ca2+-entry. Without the mechanical stimulation, an elevation of ext racellular Ca2+ cannot induce a Ca2+ transient (43). No current can be detected during reapplication of Ca2+. Under the chosen buffering conditions for [Ca2+]i (0.1 mM EGTA in the pipette solution), Ca2+ currents which are activated by store depletion, are in the range of 0.05 pA/pF at û40 mV and thus are difficult to detect (10, 12, 13, 31, 44).
[Ca2+]i Change
Pattern of [Ca2+]i Change
Figure 2 shows an example of a typical Ca2+ pattern after stimulation of an EA endothelial cell (a permanent cell line derived from human umbilical vein endothelial cells) with two different concentrations of UTP. This compound mimics the effects of ATP and binds to the P2Y2 (P2U) receptors present in these cells (50). Other agonists, like histamine and bradykinine induced similar responses. At low concentrations of the agonist, typical oscillations of [Ca2+]i which mirrors in-phase oscillations of the membrane potential but only appear in a small window of agonist concentrations. At higher concentrations of the vasoactive agonists, a typical plateau-like response in [Ca2+]i can be observed together with a hyperpolarization. Both, plateau Ca2+ and hyperploarization are more pronounced when the concentration of the agonist is increased. The plateau phase strongly depe nds on the presence of extracellular Ca2+. Withdrawal from Ca2+ in the extracellular induced a decline of [Ca2+]i and the Ca2+-plateau disappears (figure 3A and B). Agonist application in a Ca2+-free medium induced a short Ca2+-peak which reflects the intracellular release of Ca2+ and after re-addition of Ca2+, [Ca2+]i rises again which indicates influx of Ca2+ (figure 3C). These experiments clearly show that the plateau-like response during agonist application is due to influx of Ca2+ form the extracellular space.
Influx of Ca2+ not only depends on the extracellular Ca2+-cocentration but also on the membrane potential, which determines the thermodynamically driving force for the Ca2+ entry. Figure 4 shows this dependence for two different endothelial cell types. Cells derived from the pulmonary artery (CPAE cells, panel A, B) are characterised by a low plateau during agonist stimulation. In contrast, another cell type derived form human umbilical vein endothelium (EA cells, panel C, D) shows an accentuated plateau. If these cells are voltage-clamped, the driving force for inwardly transported Ca2+ can be adjusted over a wide voltage range. More negative potentials clearly increase [Ca2+]i, reduction of the driving force results in a decrease of [Ca2+]i. This dependence provides an efficient possibility for a fine tuning of the inwardly transported amount of Ca2+during stimulation of endothelial cells.
Ca2+ Influx Pathways
During stimulation of endothelial cells, influx of Ca2+ is one of the most important sources for an elevation of [Ca2+]i . Multiple entry pathways for Ca2+ exist in ECs (for a review 28, 30, 41). Recently, two pathways have been described which might be of special interest for both agonist-induced - and mechanically-induced Ca2+ entry.
Non-selective cation channels
Mechanical stimulation of endothelium activates a non-selective cation channel with a conductance of 34 pS for monovalents and 6 pS for Ca2+ which thus provides an influx pathway for Ca2+ (24, 25). Another non-selective cation channel, with a conductance of 44 pS for monovalents, is also permeable for Ca2+ and is itself activated by intracellular Ca2+ (1).
We have described Ca2+-influx pathways which are related to a non-selective cation channel which is Ca2+ permeable and probably also activated by Ca2+ itself (27, 30, 31, 38, 39). Figure 5 gives an example for whole cell currents. In EA cells, but not in CPAE ells, administration of an agonist activates a currents which reverses at potentials between +5 and +10 mV (fig.5A). This current is slowly activated and has been only observed in the presence of [Ca2+]i . Buffering with 10 mM BAPTA always completely prevented activation of this current. However, loading the cells with Ca2+ via the patch pipette is not sufficient to activate the current. Fast substitution of extracellular NaCl by NMDG-Cl induces a current, which reversed close to the theoretical K+-equilibrium potential, EK. These experiments indicate that both Na+ and K+ permeate the channel. An increase of extracellular Ca2+ induc ed a reduction of the current but also a small shift towards more positive potential indicating the Ca2+ also permeate through the channel, however, with a reduced permeability (27, 31, 39). Interestingly, application of a store-depleting inhibitor of SERCa-pumps, such as tBHQ (tert-butyl-benzohydrochinone) or thapsigargin, can also activate this NSC (figure 5B). In EA and in HUVEC cells but not in CPAE cells, the influx of Ca2+ through this NSC is strikingly coupled to agonist stimulation and depends on Ins(1,4,5)P3 production. Block of PLC with U73122 û a pyrrole-dione derivative - rapidly inhibits the Ca2+-influx whereas the pyrrolidine-dione derivative U73343, which does not inhibit PLC, is ineffective. Also NPPB, Ni2+, ecanozole, and SKF 96365 inhibit the agonist induced Ca2+-entry (Kamouchi, Viana, Nilius, unpublished). In CPAE cell, however, U73122 does not inhibit Ca2+-influx during agonist stimulation indicating that a diffe rent mechanism for the generation of the Ca2+ plateau is activated in these cells (Kamouchi, Nilius, unpublished). The mechanism of activation of this NSC is not yet completely understood. Ca2+ plays obviously the role of a co-factor. Ins(1,4,5)P3 production seems to be necessary and store depletion may provide an additional signal for channel activation. Table 1 summarises the properties of this NSC.
CRAC entry channels
Another pathway influx was first discovered in mast cells (15, 16, 45) and later on also indirectly described in endothelium (12, 13, 44). This pathway is probably much tighter coupled to depletion of intracellular Ca2+-stores as the NSC channel. It is therefore termed, Ca2+-release-activated Ca2+ current, CRAC (15, 16). It has been debated for a long time whether ECs have the typical CRAC.
Figure 6 shows a representative example for such a CRAC current in CPAE cells. Typically for such experiments, intracellular Ca2+ is strongly buffered with 10 mM BAPTA. Inclusion of Ins(1,4,5)P3 in the patch pipette, extracellular application of ionomycin, or administration of the SERCa blockers thapsigargin and tBHQ slowly activate a tiny inward current (fig.6A). Therefore, the signal(s) which is (are) activated under these conditions must be related to depletion of intracellular Ca2+ stores. At 0 mV, the current is only in the range of some pA. An elevation of extracellular Ca2+ increases the current. Removal of extracellular divalent ions induced a large, inactivating current that is blocked by micromolar concentrations of lanthanum (fig.6A). As measured from voltage ramps in the presence of extracellular Ca2+, the current reversed at very positive potentials indicating that the main charge carrier is Ca2+ (fig.6B). In the absence of Ca2+ , the current is carried by monovalents. This current is much larger than the Ca2+ current, reverses at potentials between 0 and 15 mV and typically shows a decay (fig.6C). This decay might be due to the reversed slow Ca2+-dependent activation of CRAC at low extracellular Ca2+ concentration, which is seen as a slow inactivation of the Na+ current through CRAC channels (22). Current density at 0 mV is between 10 and 20 times smaller as reported for Jurkat cells and peritoneal mast cells (10). This endothelial current shares all the now well-described properties of the typical CRAC current present in a variety of non-excitable cells (15, 45, 46).
TRP3 û a Ca2+ entry channel ?
Recently a gene family has been described which might be related to Ca2+ -entry (4, 52). Members of this so-called trp-family (from "transient receptor potential" discovered in photoreceptors of drosophila) are supposed to form functional Ca2+ channels, which can be activated by store depletion. This family is expanding (2,3, 4, 46, 52). In all isoforms three ankyrin motifs are conserved in the N-terminus. All trp┤s are characterised by six transmembrane spanning helixes and a putative pore region between helix TM5 and TM6. TM4 is not charged as it is typical for voltage-gated ion channels (2, 4, 46, 52).
We have now identified for the first time members of the trp-family in humans EC by RT-PCR techniques. Primers where designed which differentiate between the isoforms. CPAE cells express trp1and 4 but not trp3. In contrast, EA cells express trp1,3, and 4 (Flockerzi, Kamouchi, Nilius, unpublished, not shown here). To directly evaluate the functional effects of the trp expression, we have transfected CPAE cells which lack trp3 with human trp3. In CPAE cells, CRAC can be activated but not the NSC. Expression of htrp3-encoded protein, TRP3, in the plasma membrane of CPAE cells was performed by using a novel transient expression method with a novel bicistronic vector pCINeo/IRES-GFP containing htrp3 cDNA (48).This vector utilises a red-shifted variant of Green Fluorescent Protein (GFP) as an in vivo cell marker. The bicistronic unit allows coexpression of the ion channel and GFP. Transfected cells acquire typically green fluorescence. TRP3 positive CPAE cells functionally express a current, which is typically not present in CPAE cells. So far, we have not observed CRAC like currents after expression of trp3-encoded proteins. However, clearly, the trp3 transfection induced a current which can also be activated by agonists and Ins(1,4,5)P3. It has similarities with the non-selective cationic currents (NSC) which we have described in non-transfected EA cells. Figure 7 shows an example for activation of a trp3 related current in CPAE cells. Likely, NSC is TRP3. Table 3 summarised properties of TRP3.
Ion Channels and Driving Force
Role of Ion Channels in Control of Driving Force For Ca or Ca2+-Entry
Ion channels play a crucial role in ECs for the fine-tuning of electro-chemical driving forces for Ca2+. Under non-stimulated conditions, the resting potential in CPAE cells is controlled by at least three ion channels: 1. An inwardly rectifying K+ channel, 2. An outwardly rectifying Cl- channel, which is also activated by cell swelling and mechano-stimulation (volume-regulated anion channel, VRAC) and 3. a non-selective cation channel which is not identical with NSC but will not described here in detail (51).
The endothelial inwardly rectifying K+ channel, IRK
One of the most important channels for the control of the resting potential in non-stimulated cells is the K+-inward rectifier, IRK. We have identified the molecular identity of the channel. It is a member of the Kir-family, Kir2.1 (21). The full length clone is also available (11). This channel is a strongly inwardly rectifying channel with a single channel conductance 30 pS. In the C-terminus a phosphorylation motive, RRESEI, is present in which S425 seems to be important for channels regulation. We have tested the possible modulation of Kir2.1 by phosphorylation. In the presence of intracellular ATP, channels activity is very stable (fig9A). The channel can be dramatically inhibited by the absence of intracellular ATP and a run-down of channel activity will be accelerated under hypoxic condition (fig.9 B, C). This is probably due to a PP2A dependent regulation because channel inhibition can be accelerated by PP2A activators (protamine) and can be inhibited by a PP2A inhibitor (okadaic acid, fig. 9D).
The endothelial volume-regulated anion channel, VRAC
VRAC channels are present under non-stimulated conditions and play a role as "house-keeping" Cl- channels in the modulation of the resting potential of ECs. If these Cl- channels are blocked, K+ channels are now the dominating channels for the resting cells and the membrane potentials is strongly hyperpolarised. Such an example is shown in figure 8. Mibefradil, which has been described as an efficient novel Ca2+ antagonist, also inhibits endothelial Cl- channels thereby inducing a strong hyperpolarization (35). This in turn might induce beneficial effects on agonist or shear stress induced Ca2+ influx. Presence of the highly non-linear inwardly rectifying K+ channel together with a Cl- channel is a sufficient condition for appearance of such a bistable membrane potential, e.g. a fast switch between potentials close to the Cl- - (ECl) or û vice versa û close to the K+ equilibrium potential (EK) (fig.8). Interestingly, sometimes dramatic hyperpolarization cannot only be induced by activation of Ca2+-depedent K+ channels, as discussed later (see also figure 1), but also very efficiently by only modest inhibition of Cl- channels. Likely, block of Cl-channels seems to be a still underestimated tool to modulate the driving force for Ca2+-entry in endothelial cells and thus Ca2+-signalling.
Ca2+-dependent channels, CaCC and BKCa
Under condition of cell stimulation which induce an increase in [Ca2+]i, mainly two types of responses can be observed. First, CPAE cells respond with only small changes in the resting potential during an increase in [Ca2+]i. These cells only activate a Ca2+-dependent Cl- channel, CaCC (36, 37, 40). Activation of CaCC induces a shift towards a mix-potential of ECl and the reversal potential of co-activated (probably small conductance ) non-selective - or Ca2+ -selective cation channels (see below).
Another channel that is important for the regulation of the driving force for Ca2+ -influx is a big conductance, Ca2+-activated K+ channel (BKCa). Activation of these channels during an increase in [Ca2+]i shifts the membrane potential towards the equilibrium potential for potassium, EK. (see also figure 2). We have characterised this channel in human EA cells. It is an approximately 230 pS channel when measured under conditions of symmetrical K+ concentrations. Figure 10 shows a measurement from a cell-attached patch. Application of the agonist induces a fast increase in the probability of the channel being open (fig.10A). Amplitude of the single channel current is 13.9 pA at a holding potential of +60 mV (fig.10 B, D). No channel activity is observed before application of ATP (fig.10C). The open probability increases with more positive potentials. The apparent Ca2+ - affinity of the channel is increased at positive potentials, and decreases at negative potentials. These properties already define an oscillator. Pharmacologically, the endothelial BKCa is blocked by charybdotoxin (IC50 approximately 50 nM), by iberiotoxin, TEA (IC50 approximately 1 mM, 100 % block at 10 mM), and quinine. Mg2+ blocks voltage-dependently from outside. Surprisingly, the endothelial BKCa is - in contrast to the smooth muscle BKCa (5) - insensitive against NO (14). From RT-PCR analysis, we have identified the channels as hslo. The structure of this channel is not completely known. At the N-terminus, six or even seven transmembrane helices have been suggested. A segment 0 in the N-terminal is probably a couplings site for ▀-subunit. The unique C-terminus contains possibly four additional helices, H7-H10. The channel has approximately 1200 amino acids (9, 49).
Interestingly, hslo is not expressed in CPAE cells. These cells respond completely different to cell activation than the EA cells shown in figure 2. In CPAE cells no hyperpolarization is observed during agonist stimulation. Cells with a rather negative membrane potential are depolarised (figure 11A). This depolarisation approaches the equilibrium potential for Cl-, ECl , and/or - due to activation of a non-selective ion channel û even more positive values (figure 11B). Ca2+ release is followed by a very low plateau (figure 11 A and B). These cells mainly control their resting potential during agonist stimulation by activation of a Cl- - and a non-selective cation channel. As described already for TRP3, we have transfected the CPAE cells with hslo. In non-transfected control CPAE cells, ATP activates CaCC (20). The most prominent current component during ATP stimulation in hslo expressing CPAE cells is BKCa. An increase in [Ca2+]i resulted in a pronounced transient hyperpolarization, which is absent in non-transfected cells. This hyperpolarization is enhanced if CaCC is blocked by niflumic acid. The sustained component of the Ca2+ response during ATP stimulation is significantly larger in hslo transfected cells than in non-transfected cells. This plateau level correlates well with the corresponding effects of ATP on the membrane potential, indicating that the expression of cloned BKCa exerts a positive feedback on Ca2+ signals in endothelial cells by counteracting the negative (depolarising) effect of a stimulation of Ca2+-activated Cl- channels (fig. 12 A, B).
Conclusion, References & Figures
This review has summarised experimental data from our laboratory. As a bottom line, Ca2+ signalling in endothelial cells depends on the co-operativity of ion channels for Ca2+ entry, Ca2+ -release - and sequestration mechanisms.
Ca2+-entry channels are crucial elements for Ca2+ signalling in ECs. Supposedly, some of these entry channels are non-selective cation channels (NSC) and are probably regulated by intracellular Ca2+ itself (Ca2+ activated NSC). Other channels might be regulated by still unknown signals (mechano-sensor channels). The molecular nature of these channels is not yet known. Functionally identified are ion channels for Ca2+ entry which are highly selective for Ca2+. These channels are preferentially activated via a store-operated mechanism (Ca2+ release activated Ca2+ channels, CRAC). The molecular nature and the mechanism of signal transduction between Ca2+-store and a plasma membrane channel are unknown. Another pathway for Ca2+ entry is via an agonist-activated non-selective but Ca2+permeable cation channels (NSC). These channels is not or only weakly store-operated. The gating mechanism com prises a co-operativity of an agonist (receptor-Gq-Ins(1,4,5)P3 signalling cascade) with intracellular Ca2+. Likely, this channel is identical with the trp3 encoded NSC.
Importantly, the driving force for Ca2+entry also regulates Ca2+ signalling. This driving force is mainly controlled by the BKCa channels (hslo), the inwardly rectifying K+ channel (Kir2.1), at least two Cl- channels (the Ca2+-actvated Cl- channel, CaCC, and the volume-regulated, house-keeping, anion channel VRAC). A background non-selective cation channels which is present in all endothelial cells is not permeable for Ca2+ but might be involved in electrogenesis. The expression pattern of ion channels in individual EC-types determines the electrical response of these cells to activation by mechanical stimuli or by chemical agonists. A high degree of variability within the vessel tree is anticipated.
Figure 13 gives a minimal model for the main mechanisms involved in Ca2+siganlling in vascular endothelial cells. At higher agonist concentrations, the fast peak is due to release of Ca2+ from inat5racellualr stores, the plateau-like response depends on Ca2+ entry via CRAC (probably first activated) and the slowly activated NSC.
Acknowledgement
We thank Cristina Fasolato, Dominique Trouet, Gunnar Buyse ,Jean Prenen and Diane Herman for continous support. This work was supported by the Flemish F.W.O. (G.0237.95), the European Commission (ERBCHBICT941137, BMH4-CT96-0602, INTAS 94-271) and the Onderzoeksraad KU Leuven.
Legends
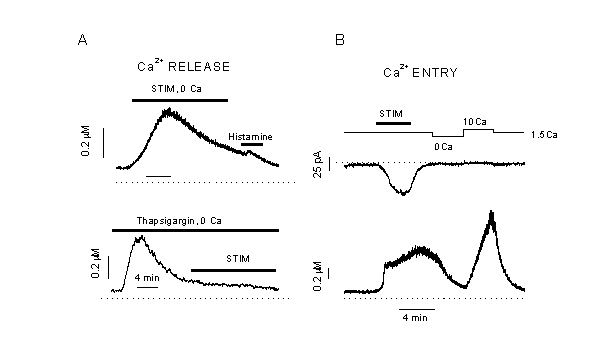 Figure 1
Figure 1
Effects of mechano-stimulation
A: Challenge of endothelial cells (human umbilical vein) by a hypotonic Ca2+-free bath solutions activates an inward current (upper trace) and induces a transient increase in [Ca2+]i (lower trace). The holding potential was -40 mV. Application of 100 M histamine did not induce a further release of Ca2+ indicating that the stores are emptied (top). HTS, applied after depletion of intracellular Ca2+-stores with 2 ÁM thapsigargin, does not evoke a Ca2+-transient (below). The same Ca2+-signals can be evoked by direct stretch or shear stress. Cell swelling by HTS is induced by changing from a 290 mOsm/l to a 185 mOsm/l Kreb┤s solution (43). Mechano-stimulation induces an intracellular release of Ca2+.
B: Swelling of endothelial cells by HTS increases [Ca2+]i and activates a Cl- current (34). The recovery of [Ca2+]i in isotonic solution is accelerated in Ca2+-free solution, but [Ca2+]i increases again after resubmission of extracellular Ca2+. Apparently, a Ca2+-entry pathway is activated by the exposure to HTS, but it is not accompanied by a significant change in transmembrane current (43).
á
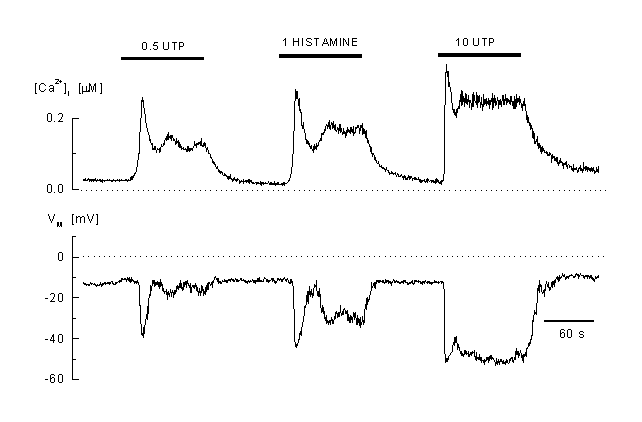
Figure 2
Calcium response to agonist stimulation
Example of the complex pattern of [Ca2+]i and membrane potential response to different agonists concentrations (0.5 microM and B: 20 microM UTP) and to 100 microM histamine (EAhy 926 cells, an umbilical vein derived cell line) These cells react to low concentrations with an oscillatory Ca2+ response which is accompanied by oscillations in the membrane potential. At high concentrations of the agonist, the response is plateau-like. A mirrored hyperpolarisation of the membrane is visible during the elevated [Ca2+]i (whole-cell current-clamp recording, the cell was pre-incubated in 2 ÁM fura-2 AM, the K-Aspartate internal solution contained 50 M fura-2) (41).
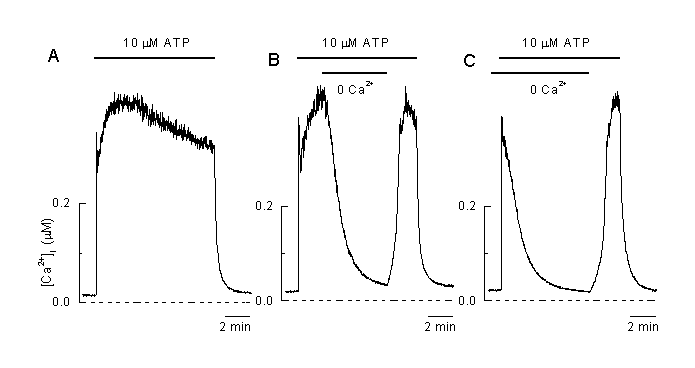
Figure 3:
The plateau phase during agonist stimulation is due to entry of Ca2+ from the extracellular solution
A: Application of 10 µM ATP induced plateau-shaped elevation of [Ca2+]i .
B: Removal of extracellular Ca2+ at the time indicated induced a decline in [Ca2+]i and a complete recovery of the [Ca2+]i plateau during the reapplication of extracellular Ca2+ in the presence of the agonist (10 µM ATP) indicating that the plateau phase is mainly due to an influx of extracellular Ca2+.
C: Application of the vasoactive agonist (10 ÁM ATP) in the absence of extracellular Ca2+ only induced a transient elevation of [Ca2+]I which mainly reflects intracellular release of Ca2+. Reapplication of extracellular Ca2+ in the presence of the agonist induced an increase in [Ca2+]I indicating that a pathway for influx of Ca2+ is still open and allows Ca2+ to flow into the cell (Viana & Nilius, unpublished).
á
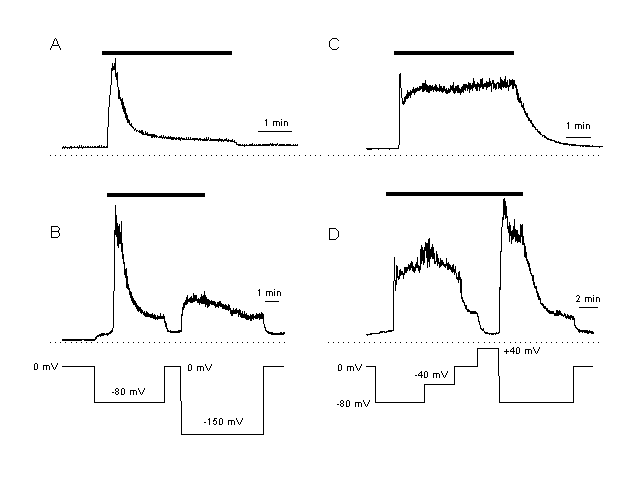
Figure 4:
Elevation of [Ca2+]i during the plateau phase depends on the inwardly
driving force for Ca2+
A, B: Ca2+ response in CPAE cells. These cells are characterised by only a small plateau response after agonist stimulation (A, response in a non-voltage clamped CPAE cell). Plateau [Ca2+]I is dependent on the driving force for Ca2+. The cells is clamped at different membrane potentials. During the plateau phase, [Ca2+]I is increased at more hyperpolarised and decreased at depolarised potentials.
C,D: Ca2+ response after the same agonist stimulation as in A, but in an EA cell. These cells respond with a much larger increase in [Ca2+]I than the CPAE cells (C, response in a non-voltage clamped cell). An increase in the driving force for Ca2+ elevated, a decrease inhibited [Ca2+]I during the plateau. These results indicate that the elevation of [Ca2+]i during the plateau phase is regulated by the driving force for Ca2+ .
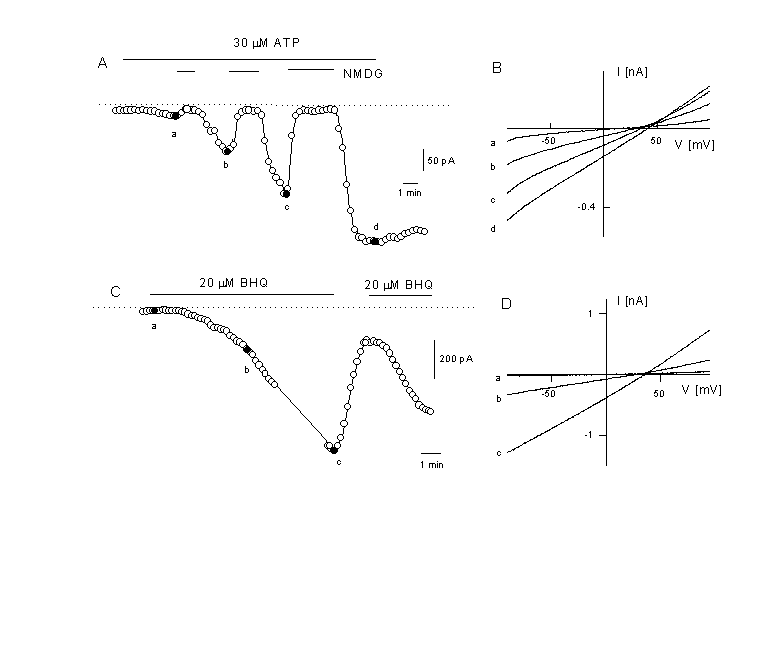
Figure 5
Activation of a non-selective cation current after agonist stimulation
A: Time course of the current activation after application of 30 ÁM ATP . Activation of the Ca2+-activated K+ current was prevented by substitution of K+ in the intracellular solution by Cs+ (extracellual K+ free). At the time indicated, NaCl was exchanged with NMDG-Cl (depicted potential is û50 mV).
B: Instantaneous IV curves were obtained from voltage ramps and depicted at the times indicated in panel A. Note the positive reversal potential between +20 and +40 mV and the disappearence of the current after removal of all cations by NMDG (N-methyl D glucamine) indicating the activation of a non-selective cation current by the agonist.
C: activation of a non-selective cation current at-50 mV in the absence of an agonist but after application of 20 µM tBHQ (a reversible SERCA pump inhibitor, tert-butylhydroquinone). Same experimental conditions as in figure 5A.
D: IV curves of the current which is activated by the store-depletion protocol. Depicted are the IV curves for obtained at the times indicated in panel C. The current is similar to the one activated by the agonist. Voltage ramps are form û100 to +100 mV (Kamouchi & Nilius, unpublished).
á
á
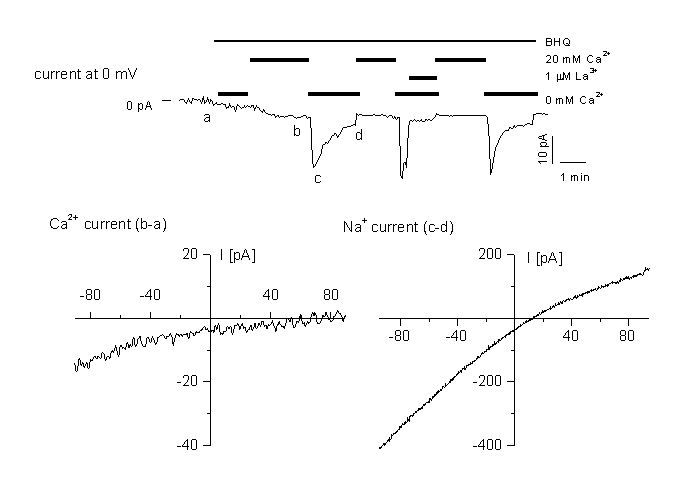
Figure 6
Activation of a store-depletion dependent current (CRAC)
A: time course of CRAC activation at 0 mV holding potential. The current is activated by application of the SERCa inhibitor BHQ. for the three different cell types. Cells were perfused with different external standard solution containing 20 mM CaCl2 or 0 mM as indicated in the figure. Cells were dialysed with the standard internal solution containing 12 mM BAPTA. Currents were blocked by 1 ÁM La3+ added to either the standard, or the Ca2+-free bath solution.
B: IV relationship in high Ca2+ medium, obtained by subtracting ramps recorded in (a) and (b).
C: IV relationship in divalent-free medium, obtained by subtracting ramps recorded in the same medium containing 1 ÁM La3+ (d) from ramps at the peak Na+ current (c). Note the block of the current by 1 ÁM LaCl3. For details, see (10).
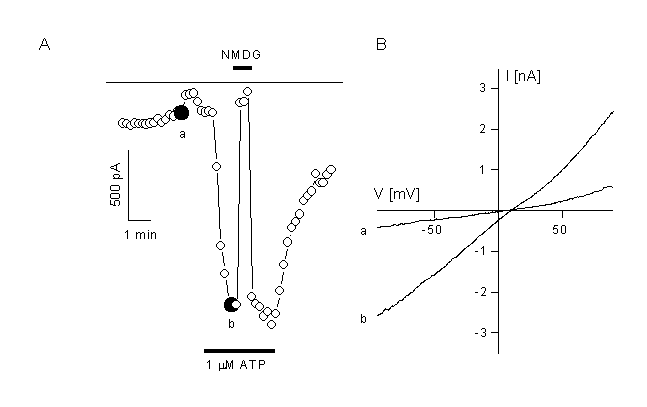
Figure 7.
Activation of a non-selective cation current in CPEA cells which are transfected with trp3.
A: CPEA cells respond to application of 1 ÁM ATP with activation of a non-selective cation channel after transfection with the trp3 cDNA. Non-transfected cells do not activate such a current. Inward current compelety disappeared after substitution of the extracellular cations with NMDG.
B. IV curves obtained form voltage ramps depicted at the times indicated in panel A.
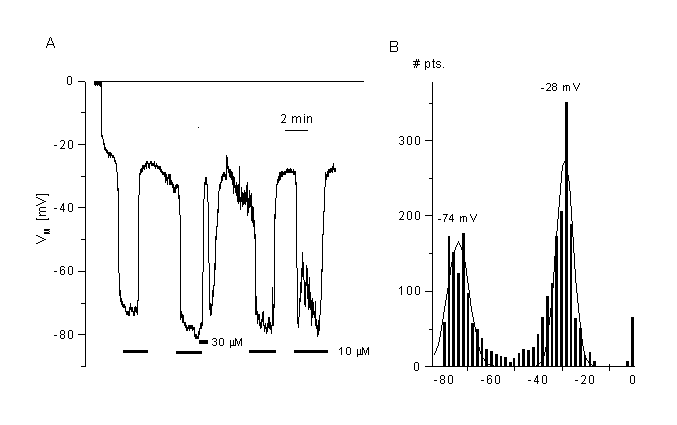
Figure 8
Effects of a chloride channel blocker (mibefradil) on the membrane potential of CPAE cells.
A. Block of Cl- channels (VRAC) induced fast hyperpolarisation. Membrane potential was measured in current clamp mode after breaking into the cell. Mibefradil (10 ÁM) induced a fast and reversible hyperpolarisation of the cells. Shown is a typical cell with low resting potential.
B. Distribution of the membrane potentials sampled at 2 Hz from the cell shown in panel A. The two peaks represent the membrane potential in the absence (control) and the presence of mibefradil. The mean values were taken from the Gaussian fits. Data obtained from the fits are: -28 mV for control, -74mV for 10 ÁM mibefradil (bin width 2 mV). Details, see (35)
á
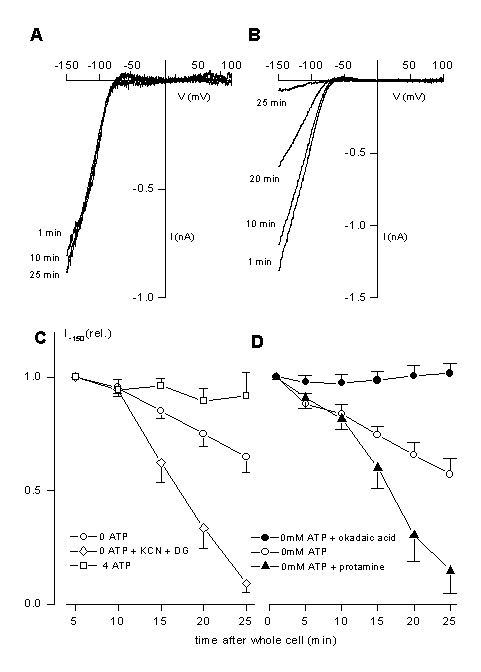 Figure 9
Figure 9
Modulation of the inwardly rectifying K+ channel, Kir2.1
A: Representative current-voltage relationships for IRK measured from voltage ramps in the presence. Currents were obtained from differences in the absence and presence of 1 mM Ba2+, which completely block Kir2.1. ATP is present in the patch pipette (4 mM). IV curves where obtained from voltage-ramps (-150 to +100 mV).
B: The same protocol as used in A but now in the absence of ATP in the patch pipette. The times are indicated which elapsed from getting whole cell access to the depicted current-voltage trace. Additionally, 2mM KCN and 5mM 2-DG were applied to the external solution immediately after the establishment of whole-cell configuration. Note the complete run-down of the current in approximately 25 minutes.
C: Run down of the activity of Kir2.1 is shown in the absence of ATP and in the absence of ATP but during application of 1 mM KCN and 5 mM 2-D-glucose (2DG). In the presence of ATP no run-down is observed.
D: Run-down of Kir2.1 can be accelerated by protamine (10 Ág ml-1), an activator of protein phosphatase PP2A. Okadaic acid (1 ÁM) completely prevented the run-down. Likely, inhibition of Kir2.1 is due to activation of PP2A. For details see also (21).
á
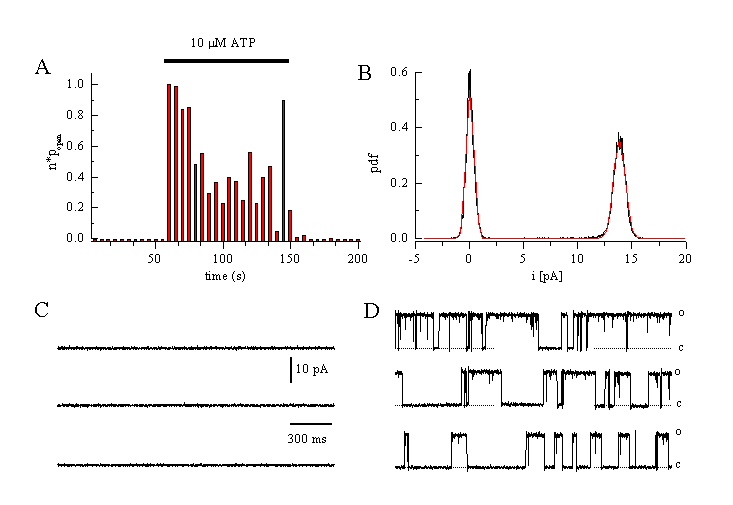
Figure 10
Single channel currents through a Ca2+-activated K+ channel evoked by ATP in EA cells.
A: Change in open probability of the BKCa-channel after application of 10 ÁM ATP (cell-attached configuration at a holding potential of +40 mV, 140 mM KCl in the patch pipette).
B: Amplitude histogram. Single channel amplitude is 13.9 pA at +60 mV indicating a channel conductance of 231 pS.
C: No channel activation is seen before ATP application.
D: Single channels traces obtained during superfusion of the cell with ATP (10 M) (Viana & Nilius, unpublished).
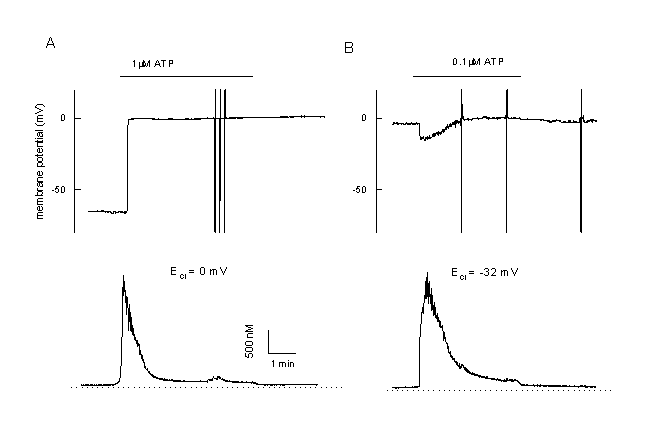
Figure 11
Calcium and membrane potential responses of bovine pulmonary artery cells (CPAE) after stimulation with an agonist
A: Simultaneous measurement of the membrane potential and [Ca2+]I in a CPAE cells during agonist stimulation (unclamped cell, loaded with Fura-2/AM, 0.1 mM EGTA in the patch pipette). The Cl- equilibrium potential is adjusted at 0 mV. During stimulation the cell is depolarised. Note the very low plateau and the low plateau of the Ca2+-signal.
B: Example of a CPAE cell, which is already depolarised. Since ECl is adjustment at more negative values, stimulation with 0.1 ÁM ATP first induces a small hyperpolarization towards ECl followed by a depolarisation. Note the completely different pattern in comparison to figure 2.
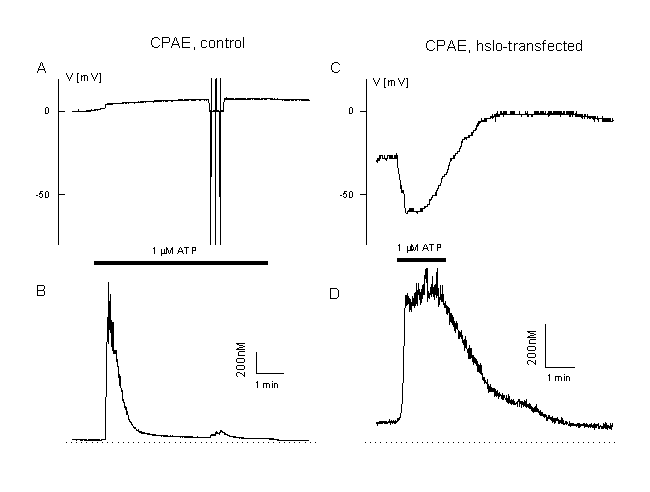
Figure 12
Calcium and membrane potential responses of bovine pulmonary artery cells after expression of the BKCa, Ca2+-activated K+ channel, hslo.
A: Measurement of the membrane potential and [Ca2+]i in a CPAE cells during agonist stimulation (unclamped cell, loaded with Fura AM, 0.1 mM EGTA in the patch pipette, 1 ÁM ATP, Cl- equilibrium potential 0 mV). Note the very low plateau and the low plateau of the Ca2+-signal and the depolarisation toward positive potentials.
B: Example of a CPAE cell which is transfected with hslo (identified by co-expression with GFP, see figure 8). Note the increased Ca2+ plateau and the large transient hyperpolarisation beyond the Cl- equilibrium potential (ECl=0 mV). More details in (20).
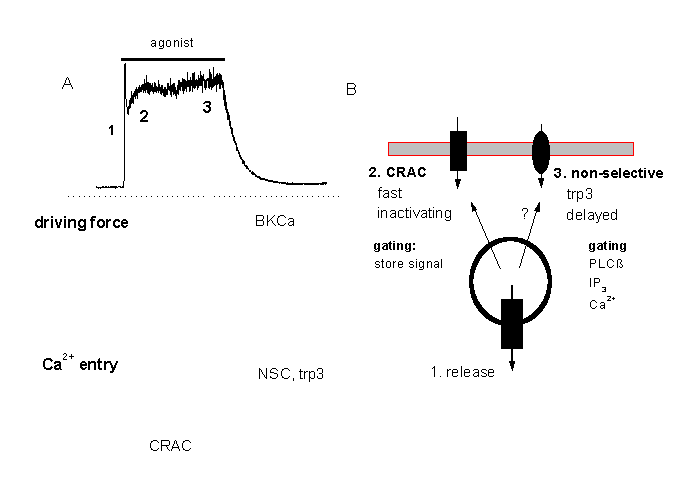
 Figure 13:
Figure 13:
A tentative minimal model for the two different modes of Ca2+ signalling in macrovascular endothelium
A: At higher agonist concentrations the Ca2+ response consists of a fast peak (corresponding o Ca2+release form intracellular stores) and a Ca2+plateau. The plateau response depends on Ca2+entry which is probably through two different pathways, CRAC and a non-selective cation channel, NSC, which is probably identical with the trp3 encoded channel. CRAC might mediate a faster influx and the delayed activated NSC.
References
- Baron, A., Frieden, M., Chabaud, F., BÚny, J.-L. 1996. Ca2+-dependent non-selective cation and potassium channels activated by bradykinin in pig coronary artery endothelial cell. Journal of Physiology 493:691-706
- Bennett, D. L., Petersen, C. C., Cheek, T. R. 1995. Calcium signalling. Cracking ICRAC in the eye. Current Biology 5:1225-8
- Berridge, M. J. 1995. Capacitative calcium entry. Biochem J 312:1-11
- Birnbaumer, L., Zhu, X., Jiang, M., Boulay, G., Peyton, M., et al. 1996. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proc Natl Acad Sci U S A 93:15195-202
- Bolotina, V. M., Najibi, S., Palacino, J. J., Pagano, P. J., Cohen, R. A. 1994. Nitric Oxide Directly Activates Calcium-Dependent Potassium Channels in Vascular Smooth Muscle. Nature 368:850-853
- Davies, P. F. 1995. Flow-mediated endothelial mechanotransduction. Physiological Reviews 75:519-560
- Davies, P. F., Barbee, K. A. 1994. Endothelial cell surface imaging: Insights into hemodynamic force transduction. News in Physiological Sciences 9153-157:157
- Davies, P. F., Tripathi, S. C. 1993. Mechanical Stress Mechanisms and the Cell - An Endothelial Paradigm. Circulation Research 72:239-245
- Dworetzky, S. I., Trojnacki, J. T., Gribkoff, V. K. 1994. Cloning and expression of a human large-conductance calcium-activated potassium channel. Mol Brain Res 27:189-93
- Fasolato, C., Nilius, B. 1998. Store depletion triggers the Calcium Release-Activated Calcium Current ICRAC in macrovascular endothelail cells: A comparison with Jurkat and embryonic kidney cells lines. Pfl³gers Archive European Journal of Physiology, in the press
- Forsyth, S. E., Hoger, A., Hoger, J. H. 1997. Molecular Cloning and Expression Of a Bovine Endothelial Inward Rectifier Potassium Channel. Febs Letters 409 (2):277-282.
- Gericke, M., Droogmans, G., Nilius, B. 1993. Thapsigargin Discharges Intracellular Calcium Stores and Induces Transmembrane Currents in Human Endothelial Cells. Pfl³gers Archiv European Journal of Physiology 422:552-557
- Gericke, M., Oike, M., Droogmans, G., Nilius, B. 1994. Inhibition of capacitative Ca2+ entry by a Cl- channel blocker in human endothelial cells. European Journal of Pharmacology 269:381-4
- Haburcak, M., Wei, L., Viana, F., Prenen, J., Droogmans, G., Nilius, B. 1997. Calcium Activated Potassium Channels In Cultured Human Endothelial Cells Are Not Directly Modulated By Nitric Oxide. Cell Calcium 21 (4):291-300.
- Hoth, M., Fasolato, C., Penner, R. 1993. Ion channels and calcium signaling in mast cells. Annals New York Academy of Sciences 707:198-209
- Hoth, M., Penner, R. 1993. Calcium Release-Activated Calcium Current in Rat Mast Cells. Journal of Physiology 465:359-386
- Inagami, T., Naruse, M., Hoover, R. 1995. Endothelium: As an endocrine organ. Annual Reviews of Physiology 57:171-189
- Jacob, R. 1990. Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. Journal of Physiology 421:55-77
- Jacob, R., Merritt, J. E., Hallam, T. J., Rink, T. J. 1988. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature 335:40-5
- Kamouchi, M., Trouet, D., De Greef, C., Droogmans, G., Eggermont, J., Nilius, B. 1997a. Functional expression of hslo Ca2+-activated K+ channels in cultured macrovascular endothelial cells. Cell Calcium 22, 497-506
- Kamouchi, M., Van den Bremt, K., Eggermont, J., Droogmans, G., Nilius, B. 1997b. Modulation of inwardly rectifying potassium channels in cultured bovine pulmonary artery endothelial cells. Journal of Physiology 504: 545-556
- Lepple Wienhues, A., Cahalan, M. D. 1996. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophysical Journal 71:787-94
- Malek, A. M., Izumo, S. 1994. Molecular aspects of signal transduction of shear stress in the endothelial cell. Journal of Hypertension 12:989-99
- Marchenko, S. M., Sage, S. O. 1996. Mechanosensitive cation channels from endothelium of excised intact rat aorta. Biophysical Journal 70:A365 (abstract).
- Marchenko, S. M., Sage, S. O. 1997. A novel mechanosensitive cationic channel from the endothelium of rat aorta. Journal of Physiology 498:419-25
- Missiaen, L., Lemaire, F. X., Parys, J. B., De Smedt, H., Sienaert, I., Casteels, R. 1996. Initiation sites for Ca2+ signals in endothelial cells. Pflugers Arch 431:318-24
- Nilius, B. 1990. Permeation Properties of a Non-Selective Cation Channel in Human Vascular Endothelial Cells. Pfl³gers Archiv European Journal of Physiology 416:609-611
- Nilius, B. 1991. Regulation of Transmembrane Calcium Fluxes in Endothelium. News in Physiological Sciences 6:110-114
- Nilius, B., Casteels, R. 1996. Biology of the vascular wall and its interaction with migratory and blood cells. In Comprehensive human physiology, ed. R. Gerger, U. Windhorst. pp. 1981-1994. Vol. 2. Berlin Heidelberg: Springer Verlag
- Nilius, B., Droogmans, G. 1995. Ion channels of Endothelial cells. In Physiology and Pathophysiology of the Heart, ed. N. Sperelakis. pp. 961-973 Kluwer Academic Publishers
- Nilius, B., Droogmans, G., Gericke, M., Schwartz, G. 1993a. Non-selective ion pathways in human endothelial cells. In Nonselective cation channels, Physiology and Biophysics, ed. D. Siemen, J. Hescheler. pp. 269-280. Basel: Birkhõuser Verlag
- Nilius, B., Eggermont, J., Voets, T., Buyse, G., Manolopoulos, V., Droogmans, G. 1997a. Properties of volume-regulated anion channels in mammalian cells. Progress in Biophysics and Molecular Biology 68: 69-119
- Nilius, B., Eggermont, J., Voets, T., Droogmans, G. 1996. Volume-activated Cl--channels. General Pharmacology 27:67-77
- Nilius, B., Oike, M., Zahradnik, I., Droogmans, G. 1994. Activation of a Cl- current by hypotonic volume increase in human endothelial cells. Journal of General Physiology 103:787-805
- Nilius, B., Prenen, J., Kamouchi, M., Viana, F., Voets, T., Droogmans, G. 1997b. Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2+- and volume-activated Cl- channels in macrovascular endothelial cells. British Journal of Pharmacology 121:547-555
- Nilius, B., Prenen, J., Szucs, G., Wei, L., Tanzi, F., et al. 1997c. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. Journal of Physiology 498, 381-396
- Nilius, B., Prenen, J., Voets, T., Vandenbremt, K., Eggermont, J., Droogmans, G. 1997d. Kinetic and Pharmacological Properties Of the Calcium Activated Chloride Current In Macrovascular Endothelial Cells. Cell Calcium 22:53-63.
- Nilius, B., Riemann, D. 1990. Ion channels in human endothelial cells. General Physiology and Biophysics 9:89-112
- Nilius, B., Schwarz, G., Oike, M., Droogmans, G. 1993b. Histamine-Activated, Non-Selective Cation Currents and Ca2+ Transients in Endothelial Cells from Human Umbilical Vein. Pfl³gers Archiv European Journal of Physiology 424:285-293
- Nilius, B., Sz³cs, G., Heinke, S., Voets, T., Droogmans, G. 1997e. Multiple Types Of Chloride Channels In Bovine Pulmonary Artery Endothelial Cells. Journal of Vascular Research 34 :220-228.
- Nilius, B., Viana, F., Droogmans, G. 1997f. Ion channels in vascular endothelium. Annual Review of Physiology 59:145-170
- Oike, M., Droogmans, G., Nilius, B. 1994a. Amplitude modulation of Ca2+ signals induced by histamine in human endothelial cells. Biochim Biophys Acta 1222:287-91
- Oike, M., Droogmans, G., Nilius, B. 1994b. Mechanosensitive Ca2+ transients in endothelial cells from human umbilical vein. Proceedings of the National Academy of Sciences of the USA 91:2940-2944
- Oike, M., Gericke, M., Droogmans, G., Nilius, B. 1994c. Calcium entry activated by store depletion in human umbilical vein endothelial cells. Cell Calcium 16:367-76
- Penner, R., Fasolato, C., Hoth, M. 1993. Calcium influx and its control by calcium release. Curr Opin Neurobiol 3:368-74
- Philipp, S., Cavalie, A., Freichel, M., Wissenbach, U., Zimmer, S., et al. 1996. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. Embo Journal 15:6166-6171
- Schwarz, G., Callewaert, G., Droogmans, G., Nilius, B. 1992. Shear stress-induced calcium transients in endothelial cells from human umbilical cord veins. Journal of Physiology 458:527-538
- Trouet, D., Nilius, B., Voets, T., Droogmans, G., Eggermont, J. 1997. Use of a bicistronic GFP-expression vector to characterise ion channels after transfection in mammalian cells. Pfl³gers Archiv European Journal of Physiology 434:632-638
- Tseng Crank, J., Foster, C. D., Krause, J. D., Mertz, R., Godinot, N., et al. 1994. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13:1315-30
- Viana, V., De Smedt, H., Droogmans, G., Nilius, B. 1998. Calcium signalling through nucleotide receptor P2Y2 in cultured human vascular endothelium. Cell Calcium 24: 117-127
- Voets, T., Droogmans, G., Nilius, B. 1996. Membrane currents and the resting membrane potential in cultured bovine pulmonary artery endothelial cells. Journal of Physiology 497:95-107
- Zhu, X., Jiang, M., Peyton, M., Boulay, G., Hurst, R., et al. 1996. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 85:661-71
Tables:
Table 1: Properties of the non-selective cation channels (NSC) in endothelial cells
Size: ~ 20pA/pF at -50 mV, not in all endothelial cells present, 25-30 pS (single channel)
Permeation: Na+ > Cs+ > Ca2+>>> NMDG; Ca2+ permeable: PCa/PNa = 0.03
Activation: agonsist (PLC dependent), Ins(1,4,5)P3, BHQ, thapsigargin
Kinetics: time and voltage-independent, but slow and delayed activation
Ca2+ : permissive Ca2+ is required, but no activation by Ca2+
Modulation: Inhibition by PLC blockers (U73122), weak inhibition by La3+, Gd3+
Table 2: Properties of calcium-release-activated Ca2+ channels (CRAC)
Probably present in all endothelial cells
Size: ~ -0.5 pA/pF at -50 mV (intracellular 12 mM BAPTA)
Permeation: Ca2+ >> Na+ > Cs+; Ca2+ >> Ba2+ in the presence of Ca2+
Kinetics: Inward rectification, inactivation
Activation: Ins(1,4,5)P3, BHQ, thapsigargin, ionomycin, no Ca2+ necessary, store-operated
Modulation: ÁM block by lanthanum, inhibition by NPPB, ecanozol, Ni2+
Table 3: Properties of non-selective cation currents induced by trp3 expression.
Size: ~ - 25 pA/pF at -50 mV, 25-30 pS (noise analysis)
Permeation: Na+ > Cs+ > Ca2+ >> NMDG; Ca2+ permeable: PCa / PNa = 0.9
Activation: agonists (PLC), Ins(1,4,5)P3, ionomycin; weak of none store -dependence
Kinetics: Fast activation by agonists
Modulation: Inhibition by PLC blockers (U73122); weak inhibition by La3+, Gd3+, Mg2+
| Discussion Board | Previous Page | Your Symposium |