Invited Symposium: Oxidative Stress and the CNS
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Oxidative stress has been implicated in playing a central role in ageing and its associated neurodegenerative diseases [2, 17, 18, 20, 24, 27, 28, 33, 35]. The reason is that oxidative stress promotes glutamate excitotoxicity that leads to uncontrollable rises in intracellular Na+ and Ca2+ that in turn leads to ATP depletion followed by cell death. These four - excitotoxicity, rises in intracellular Ca2+, oxidative stress and ATP depletion - form a vicious interactive spiral that results in cell death. One could attempt to prevent the formation of such an interactive spiral by interfering with the development of any one of the components of this spiral. Attempts to control excitotoxicity or abnormal rises in intracellular Ca2+ interferes with the normal function of glutamate and Ca2+ and, hence, this approach has had limited therapeutic efficacy, e.g., [23]. Minimizing oxidative stress would appear to be the most promising means of preventing the establishment of the above vicious spiral to cell death. The attempts to prevent the development of oxidative stress also has had minimal therapeutic efficacy. The reason for this is that too little appreciation has been given to the central role that peroxides have in the development of oxidative stress.
Strong Oxidant Production & Scavenging
A brief outline of strong oxidant production is given below. For a more detailed review see [10, 16] where all the appropriate references are also given. The major cellular sources of free radicals under normal physiological conditions are the mitochondria where ~3% of oxygen is incompletely reduced to the superoxide anion. The CNS which comprises about 2% of the body’s mass consumes, when the body is at rest, about 20% of all the oxygen. Hence, there is a large production of superoxide anion by CNS tissue; furthermore, superoxide production by brain increases as one ages [26]. Although relatively innocuous, superoxide can give rise to strong oxidants such as singlet oxygen and interact with other relatively innocuous compounds such as nitric oxide radical or hydrogen peroxide to give rise to the hydroxyl radical, nitrogen dioxide as outlined in Figure 1.
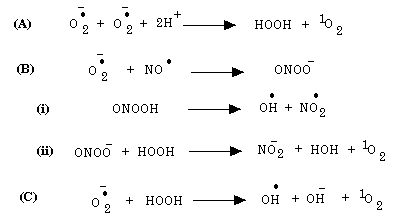 Fig. 1: Strong Oxidants Produced From Superoxide Anion.
Fig. 1: Strong Oxidants Produced From Superoxide Anion.
These strong oxidants can oxidize proteins, especially thiol proteins, ribonucleic acids as well as polyunsaturated fatty acids. Hence, an important free radical scavenging action is that mediated by superoxide dismutase (SOD), where superoxide is converted to hydrogen peroxide and molecular oxygen as indicated in the following reaction indicated by Figure 2.
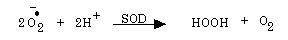 Fig. 2: Superoxide Dismutase Action.
Fig. 2: Superoxide Dismutase Action.
There are three SOD isoforms: i) an extracellular Cu,Zn-SOD, a cytosolic Cu,Zn-SOD and a mitochondrial Mn-SOD [9]. The hydrogen peroxide produced is not innocuous since it can be converted to the strong oxidant, the hydroxyl radical, by transition metal ions as indicated in Figure 3.
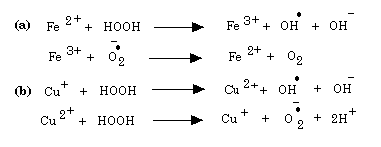 Fig. 3: Transition Metal Ions And Production Of Strong Oxidants.
Fig. 3: Transition Metal Ions And Production Of Strong Oxidants.
Note that superoxide and hydrogen peroxide are involved in the redox cycling of iron and copper, respectively. The hydroxyl radical is a powerful oxidant and can extract an electron from another molecule or can hydroxylate another molecule. Two major means by which the hydroxyl radical can cause cell damage is by causing changes in DNA leading to mutations [3] and by the initiation of a chain of peroxidations of polyunsaturated fatty acids [10]. One should keep in mind that transition metal cations tend to be localized to anionic structures such as the phosphate groups of phospholipids and DNA, a consequence of which is that the generation of hydroxyl radicals tends to be in regions where they can cause the most damage. Figure 4 illustrates how lipid peroxidation chains can be initiated and how they can be prevented. LH represents a polyunsaturated fatty acid, LOOH a lipid hydroperoxide, LOO a lipid peroxyl radical, LO a lipid alkoxyl radical, TOH vitamin E and AscH ascorbic acid.
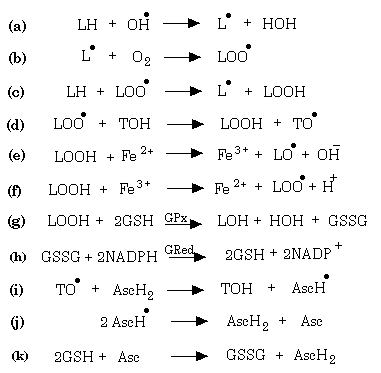 Fig. 4: Production and Scavenging of Lipid Peroxides.
Fig. 4: Production and Scavenging of Lipid Peroxides.
Note in equations ‘a’ to ‘c’ the initiation of a chain reaction of lipid peroxidations that can only be stopped by either two lipid radicals interacting or by the inactivation of a lipid radical by vitamin E. Inactivation of a lipid peroxyl radical results in the formation of a lipid peroxide and a vitamin E radical. The lipid peroxides can interact with either iron-II or iron-III and give rise to alkoxyl and peroxyl radicals, respectively, each of which can initiate new chains of lipid peroxidation. Peroxidized plasmalemmal, endoplasmic reticular, mitochondrial membrane lipids greatly interfere with cell function. It is vital for the cell to scavenge the peroxides produced. It does this principally using the selenoenzyme glutathione peroxidase (GPx) which in turn uses reduced-glutathione (GSH) as the electron donor as indicated in equation ‘g’ [7]. There are several members of the GPx family: i) a plasma form whose function is unclear since there is very little GSH in plasma [29], ii) a cytosolic and mitochondrial form (GPx1) [4, 7], iii) a membrane-associated phospholipid hydroperoxide glutathione peroxidase (GPx4) [32], and a gastrointestinal glutathione peroxidase (GPx-GI) [5]. Of significance is that GPx activities in brain decreases with age [34]. Oxidized-glutathione (GSSG) is reduced by glutathione reductase using NADPH as the source of electrons. GPx can scavenge both hydrogen peroxide as well as organic peroxides and has a high affinity for such peroxides. Catalase, on the other hand, can scavenge only hydrogen peroxide and has a low affinity for hydrogen peroxide. The efficiency by which GPx can scavenge peroxides increases with increasing GSH concentration [6, 25, 32] (note Figure 5). In other words, relatively small increases in GSH concentration has a marked effect on the ability of GPx to scavenge peroxides. Indeed, increasing intracellular GSH has been demonstrated to increase the ability of cells to scavenge strong oxidants [30, 31], note Figure 5. Conversely decreasing intracellular GSH results in greater damage following oxidative stress [11, 13, 19, 22]. GSH is also important in the regeneration of ascorbate which has been used to reduce the vitamin E radical back to vitamin E. Hence, GSH plays a very central role in the ability of cells to manage oxidative stress [14-16].
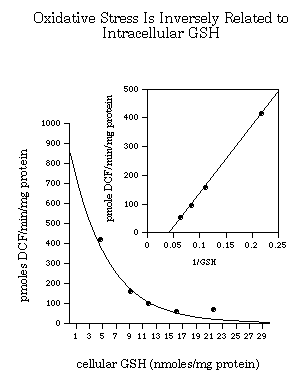 Fig. 5: Oxidative Stress Is Inversely Related to Intracellular GSH.
Fig. 5: Oxidative Stress Is Inversely Related to Intracellular GSH.
In Figure 5, taken from [30], the amount of strong oxidant (such as the hydroxyl radical) being formed is measured by the oxidation of the non-fluorescent DCFH to DCF under a perturbation that causes increased hydrogen peroxide formation. In this experiment hydrogen peroxide was caused to form by exposing oligodendrocyte precursors to blue light (480 nm at 140 mW/m2): this causes excitation of compounds such as riboflavin which in turn donates an electron to molecular oxygen resulting in formation of superoxide that in turn is dismutated to hydrogen peroxide. Intracellular GSH was manipulated in these cells from the ~5 nmoles/mg protein normally found in these cells to ~22 nmoles/mg protein, the level that is normally found in astrocytes. Note that a relatively modest increase in GSH from 5 nmoles/mg protein to 7 nmoles/mg protein can halve the oxidative stress experienced by these cells.
GSH and Cell Stress
GSH is synthesized according to the following two reactions in Figure 6.
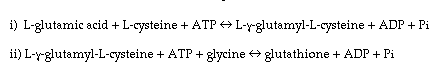 Fig. 6: GSH Synthesis.
Fig. 6: GSH Synthesis.
Glutamyl-cysteine synthase (GCS: reaction ‘i’) is the rate-limiting enzyme in regulating GSH levels [21]. It is a heterodimer comprised of a 73 kD heavy subunit and a 27.7 kD light subunit. The activity of glutamyl-cysteine synthase, the key enzyme in GSH synthesis drops with age [12]. Another important enzyme regulating the intracellular levels of GSH is glutathione reductase, which uses NADPH as the electron donor to reduce oxidized-glutathione to reduced-glutathione. Cerebral GSH levels has been demonstrated to decrease while GSSG levels rise with age [12]. GSH is not only important for the scavenging of peroxides but also for the reduction of oxidized-ascorbic acid which is necessary for the regeneration of vitamin E. GSH also plays an important role in determining how readily proinflammatory genes can be activated since GSH acts as the intracellular redox buffer. High GSH:GSSG ratios tend to inhibit the activation of the redox-sensitive transcription factor NF-kappaB [8]. NF-kappaB is the key transcription factor involved in activating transcription of pro-inflammatory genes such as cyclo-oxygenase-2, inducible nitric oxide synthase, intercellular adhesion molecules and proinflammatory cytokines [1].
Concluding Remarks
Most neurodegenerative diseases become more common as we age. There are a number of known reasons for this, including increased inefficiencies in mitochondrial function leading to increased superoxide production, decreased abilities to produce GSH and to reduce oxidized-glutathione to GSH, decreased activities of GPx, etc. It is my thesis that raising GSH in neural and endothelial cells will inhibit the formation of strong oxidants and thus delay the onset of a variety of neurodegenerative diseases. Increases in tissue GSH can be accomplished either by administering compounds that increase intracellular levels of the rate-limiting amino acid, cysteine, for GSH synthesis or by promoting the activity of the rate-limiting enzyme, glutamyl-cysteine synthase. Administration of the cysteine pro-drug, N-acetylcysteine has been demonstrated to result in better survival of motoneurons in a mouse model of motoneuron disease (see paper by Henderson and Roder in this Symposium). One of the research directions in which my laboratory is interested is raising intracellular GSH by promoting the activity of glutamyl-cysteine synthase.
References
- Baldwin, A.S., The NF-kappa b and i kappa b proteins: new discoveries and insights, Annu Rev Immunol, 14 (1996) 649-683.
- Beal, M.F., Aging, energy, and oxidative stress in neurodegenerative diseases, Ann Neurol, 38 (1995) 357-366.
- Breen, A.P. and Murphy J.A., Reactions of oxyl radicals with DNA, Free Rad Biol Med, 18 (1995) 1033-1077.
- Cheng, W.-H., Ho Y.-S., Ross D.A., Valentine B.A., Combs G.F. and Lei X.G., Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidase in various tissues, J Nutr, 127 (1997) 1445-1450.
- Chu, F.-F., Doroshow J.H. and Esworthy R.S., Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI, J Biol Chem, 268 (1993) 2571-2576.
- Esworthy, R.S., Chu F.-F., Geiger P., Girotti A.W. and Doroshow J.H., Reactivity of plasma glutathione peroxidase with hydroperoxide substrates and glutathione, Arch Biochem Biophys, 307 (1993) 29-34.
- Flohé, L., Glutathione peroxidase: fact and fiction. In I. Fridovich (Ed.), Oxygen Free Radicals and Tissue Damage, Ciba Foundation Symposium 65, Excerpta Medica, Amsterdam, 1979, pp. 95-113.
- Flohé, L., Brigelius-Flohe R., Saliou C., Traber M.G. and Packer L., Redox regulation of NF-kappa B activation, Free Radic Biol Med, 22 (1997) 1115-1126.
- Fridovich, I., Superoxide radical and superoxide dismutases, Ann Rev Biochem, 64 (1995) 97-112.
- Halliwell, B. and Gutteridge J.M.C., Free Radicals in Biology and Medicine, Second edn., Clarendon Press, Oxford, 1989, 543 pp.
- Huang, J. and Philbert M.A., Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis, Brain Res, 711 (1996) 184-192.
- Iantomasi, T., Favilli F., Marraccini P., Stio M., Treves C., Quatrone A., et al., Age and GSH metabolism in rat cerebral cortex, as related to oxidative and energy parameters, Mech Ageing Develop, 70 (1993) 65-82.
- Jain, A., Mĺrtensson J., Stole E., Auld P.A.M. and Meister A., Glutathione deficiency leads to mitochondrial damage in brain, Proc Natl Acad Sci USA, 88 (1991) 1913-1917.
- Juurlink, B.H.J., Central role of glutathione in governing the response of astroglial and oligodendroglial cells to ischemia-related insults, Recent Res Develop Neurochem, 1 (1996) 179-192.
- Juurlink, B.H.J., Response of glial cells to ischemia: roles of reactive oxygen species and glutathione, Neurosci Biobehav Rev, 21 (1997) 151-166.
- Juurlink, B.H.J. and Paterson P.G., Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and management strategies, J Spinal Cord Med, in press (1998).
- LeBel, C.P. and Bondy S.C., Oxidative damage and cerebral aging, Prog Neurobiol, 38 (1992) 601-609.
- Markesbery, W.R., Oxidative Stress Hypothesis In Alzheimers Disease, Free Radical Biology And Medicine, 23 (1997) 134-147.
- Mĺrtensson, J., Jain A., Stole E., Frayer W., Auld P.A. and Meister A., Inhibition of glutathione synthesis in the newborn rat: a model for endogenously produced oxidative stress, Proc Natl Acad Sci USA, 88 (1991) 9360-9364.
- Mattson, M.P., Mark R.J., Furukawa K. and Bruce A.J., Disruption of brain cell ion homeostasis in Alzheimer's disease by oxy radicals, and signalling pathways that protect therefrom, Chem Res Toxicol, 10 (1997) 507-517.
- Meister, A., Metabolism and function of glutathione. In D. Dolphin, O. Avramovic´ and R. Poulson (Eds.), Glutathione. Chemical, Biochemical, and Medical Aspects, Vol. A, John Wiley & Sons, New York, 1989, pp. 367-474.
- Mizui, T., Kinouchi H. and Chan P.K., Depletion of brain glutathione by butathione sulfoxamine enhances cerebral ischemic injury in rats., Am J Physiol, 262 (1992) H313-H317.
- Muir, K.W. and Lees K.R., Clinical experience with excitatory amino acid antagonist drugs, Stroke, 26 (1995) 503-513.
- Olanow, C.W., A radical hypothesis for neurodegeneration, Trends Neurosci, 16 (1993) 439-444.
- Paglia, D.E. and Valentine W.M., Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase, J Lab Clin Med, 70 (1967) 158-169.
- Sawada, M. and Carlson J.C., Change in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat, Mech Ageing Develop, 41 (1987) 125-137.
- Simonian, N.A. and Coyle J.T., Oxidative stress in neurodegenerative diseases, Annu Rev Pharmacol Toxicol, 36 (1996) 83-106.
- Stadtman, E.R., Protein oxidation and aging, Science, 257 (1992) 1220-1224.
- Takahashi, K., Avissar N., Whitin J. and Cohen H., Purification and characterization of human plasma glutathione peroxidase: a selenoglycoprotein distinct from the known cellular form, Arch Biochem Biophys, 256 (1987) 677-686.
- Thorburne, S.K. and Juurlink B.H.J., Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress, J Neurochem, 67 (1996) 1014-1022.
- Tsan, M.-F., Danis E.H., Del Vecchio P.J. and Rosano C., Enhancement of intracellular glutathione protects endothelial cells against oxidant damage, Biochem Biophys Res Comm, 127 (1985) 270-276.
- Ursini, F., Maiorino M. and Gregolin C., The selenoenzyme phospholipid hydroperoxide glutathione peroxidase, Biochim Biophys Acta, 839 (1985) 62-70.
- Yim, M.B., Kang J.-H., Yim H.-S., Kwak H.-S., Chock P.B. and Stadtman E.R., A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide, Proc Natl Acad Sci USA, 93 (1996) 5709-5714.
- Zhang, L.P., Maiorino M., Roveri A. and Ursini F., Phospholipid hydroperoxide glutathione peroxidase: specific activity in tissues of rats of different age and comparison with other glutathione peroxidases, Biochim Biophys Acta, 1006 (1989) 140-143.
- Zorov, D.B., Mitochondrial damage as a source of diseases and aging: a strategy of how to fight these, Biochim Biophys Acta, 1275 (1996) 10-15.
| Discussion Board | Previous Page | Your Symposium |