Invited Symposium: Molecular Mechanisms of Ageing
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
The Senescence-Accelerated Mouse (SAM)
Development of the SAM strain (Senescence-Accelerated Mouse)
The SAM strain of mice has been derived from AKR/J strain which were donated by the Jackson Laboratory (Bar Harbor, Maine, USA) and maintained in our laboratory. Certain littermates which became senile at an early age in life and had a shorter life span were noticed. In 1975, five litters with early senescence were selected as the progenitors of the SAMP. Three litters in which the aging process seemed normal were also selected as the progenitors of SAMR [1-4]. Thereafter, retrospective pedigree selection and inbreeding [5] were applied based on the degree of senescence [1,3,6,7], the lifespan and the age-associated pathologic phenotypes. These mice were named the Senescence-Accelerated Mouse (SAM) in 1981 [1] (Fig.1)
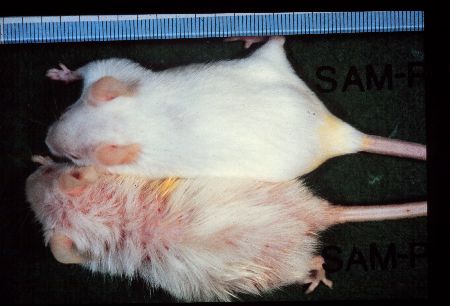 Fig1: SAMP1 (lower) and SAMR1 (upper) males of 12-month-old.
Fig1: SAMP1 (lower) and SAMR1 (upper) males of 12-month-old.
A possible mechanism which developed the SAM strain
The genetic study revealed that there is at least one genotype in each SAM strain that differs from that in the AKR/J strain and in some loci, three haplotypes have been recognized [3,8]. The results suggest that a recombination of the genes from the gene pools is assumed to have occurred successively during the selective inbreeding after unexpected outbreeding (Fig.2). At the present time, the SAM strain can be considered as a group of related inbred strains.
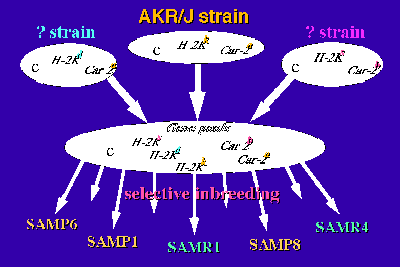 Fig2: The generation of the SAM strains.
Fig2: The generation of the SAM strains.
Senescence Acceleration in SAM Mice
Aging process in SAM mice
The senescence process of SAMP mice seems to be accelerated after normal development and maturation (Fig.3).
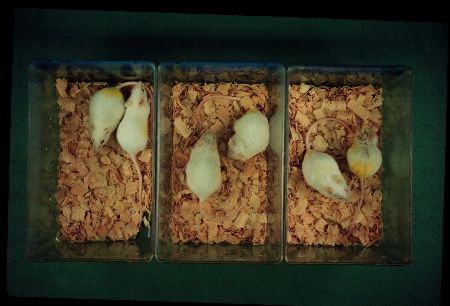 Fig.: From left to right, there are three pairs of SAMP11 and SAMR1 males of 3, 6 and 12 months of age. Both strains can not be distinguished before 6-month-old.
Fig.: From left to right, there are three pairs of SAMP11 and SAMR1 males of 3, 6 and 12 months of age. Both strains can not be distinguished before 6-month-old.
The survivorship curve and Gompertzian function indicated that the increase in the age specific death rate of the SAMP was greater as compared to those in the SAMR [1]. Because of a steeper increase in the degree of senescence [1,3,6,7] in SAMP, the difference between the two strains maintained in our colony maintained under conventional condition became obvious after 6 months of age [6]. Although there were some differences in the frequency of relatively causes of death in each strain, there were no strain specific causes of death between SAMP and SAMR [1, 9].
In both SAMP1 and SAMR1, the body weight increased rapidly up to 14 weeks of age, and there were no differences between either strain in both males and females [1]. The time at which reproductive capability was reached in the SAMP3, SAMP11 and SAMR1 was between 39 to 45 days after birth and there was no difference
Concept of accelerated senescence
In our studies, the words "aging" and "senescence" have been used in a different context. Aging consists of the biological phenomena observed in an individual organism from the time of fertilization to the time of death. Strehler [10] proposed 4 criteria: universality, intrinsicality, time dependence and deleteriousness, for delineating age-related changes. Based on this concept, we consider the first three are criteria which delineate "aging", whereas all 4 criteria together delineate "senescence". In this context, "senescence" can be considered to be an aging phenomenon which occurs after maturation. Conceptually, there can be three kinds of senescence processes: "normal senescence", "accelerated senescence" and "premature senescence or premature aging" (Fig.4).
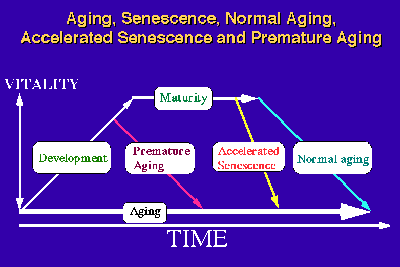 Fig.: A concept of accelerated senescence.
Fig.: A concept of accelerated senescence.
The latter two senescence processes become apparent only when they are compared to the senescence process which we can consider as "natural or normal". Although "accelerated senescence" and "premature aging" can be clearly defined conceptually, these processes typically overlap. The aging process in "premature aging" has an earlier time of onset as well as an accelerated tempo and an exacerbation of age-associated pathology [11]. In the SAMP strains, normal development and maturity of reproductive function was observed, and the values for many (but not all) physiological and pathological parameters are the same as those in the SAMR strains at young to mature age. Due to the rapid progression of the senescence process, the median survival time of each SAMP strain (211 - 415 days) was about 37-73 % of the corresponding of SAMR1 strain (568 days) and the degree of senescence at 8 months of age in the SAMP strains (5.2 - 11.7) was about 1.5-3.5 times higher than in the SAMR1 strain (3.4) under our conventional rearing conditions [3]. This observation indicates that the senescence process in SAMP is more characteristic of "accelerated senescence" than of "premature aging".
Although the median survivor time of SAMR1 in our animal facility is 568 days, those of the SAMR1 reared under specific pathogen free conditions are extended to 1.3 - 1.5 times of that of conventionally reared mice (personal communication, Abe, T. et al.). This value corresponds to those of common, long-lived strains, including C57BL and CBA [12].
Senescence acceleration in vitro
The senescence acceleration is observed in not only an individual organism but also in the cells isolated from body and grow in vitro. Cultured fibroblasts derived from SAMP11 neonates [13] or SAMP6 adults [14] had accelerated in vitro aging and reduced lifespans in culture. The observation of the acceleration of changes in DNA ploidy of these fibroblasts with in vitro aging suggests that the cell lines of these SAMP mice might have higher generation of, and/or susceptibility to, oxidative stress [15].
Age-associated Pathologies in SAM Mice
Systematic studies of various age-associated pathologies of SAM mice revealed that these pathologies could be assessed as models for several age-associated disorders observed in humans (Fig.5).
 Fig5:Age-associated pathologies observed in SAMP strains.
Fig5:Age-associated pathologies observed in SAMP strains.
Senile osteoporosis (SAMP6)
Since spontaneous leg fracture in a few aged SAM mice has been observed, we began to systematically examine the age-related changes in bone in the SAM strains, and found that SAMP6 showed senile osteoporosis characterized by a low peak bone mass at their maturation [16]. A decrease in bone formation due to a paucity of osteoblast progenitor cells [17,18], and an increase in bone resorption due to the enhanced maturation of osteoclast cells has been suggested [19] as the causes of low peak bone mass. Cross-mating studies with SAMP2 (shows highest peak bone mass) indicated the low peak bone mass of the SAMP6 was controlled polygenically by a relatively small number of genes [20]. Our recent whole genome scan for QTLs for specifying peak bone mass indicated three loci, on chromosome 11, on chromosome 13 and on X chromosome [21]
Age-related deficits in learning and memory (SAMP8, SAMP10)
The characteristic neurobiological senescent phenotype, age-related deficits in learning and memory, was reported in SAMP8 (SAMP8/Ta [22], SAMP8 [23]) and SAMP10 (SAMP10//Ta [24], SAMP10 [25]). SAMP8 mice showed impairments in passive avoidance tasks [22,23], one-way [22], T-maze [26] and Sidman [27] active avoidance tasks and an impairment of their spatial memory task ability [24]. They had age-related emotional disorders characterized by reduced anxiety-like behaviour [28]. SAMP10 had mildly impaired passive avoidance task [24] and active avoidance response [24,25], as seen in SAMP8. Impairment in the active avoidance task in SAMP10 could be attributed to behavioral depression [24]. The SAMP8 and SAMP10 showed a profound disorder of their circadian rhythms of spontaneous motor activity and drinking behaviors [24].
Histopathology
The SAMP8 showed age-related appearances of spongiform degeneration in the brain stem [29], and of PAS-positive granular structures in their hippocampal formation [30], and astrogliosis in their brain stem [29], hippocampus, pyriform cortex, brain stem nuclei and white matter [31]. Clusters of activated microglia were also seen around the vacuoles in the brain stem [32]. A monoamine-oxidase-B-positive granular structure was found in hippocampus of old mice [33]. Beta/A4 protein-like immunoreactive granular structures were observed in various regions, including the medial septum, cerebral cortex, hippocampus, cerebellum, and some cranial nerve nuclei and roots and increased markedly in number with age [34]. Other age-dependent histological changes included cortical atrophy in the pyriform cortex, neuronal cell loss in the locus ceruleus and lateral tegmental nucleus, intraneuronal accumulation of lipopigment in Purkinje cells, and eosinophilic inclusion bodies in thalamic neurons [31,35]. Similar changes were also observed to a lesser degree in SAMP10. However, the most characteristic age-related change in SAMP10 was brain atrophy, particularly in the frontal portion of the cerebrum, frontal, parietal, temporal and occipital cerebral cortices, especially olfactory cortex and amygdala [36]. The large neurons in these areas typically shrank and/or disappeared in aged SAMP10 [36]. An age-related decline in nerve growth factor (NGF) immunoreactivity was also found in the substantia innominata, but retained in each layer of the neocortex [37].
Blood-brain barrier function was impaired with advancing age in the olfactory bulb and medial hippocampus in SAMP8 [38-41].
Neurobiochemical studies
Muscarinic acetylcholine receptors, alpha 2-adrenoreceptors, N-methyl-D-aspartate (NMDA) receptor channels and L-type Ca2+channels were all changed in the cerebral cortex and hippocampus in aged SAMP8 [42]. High levels of K+ - and NMDA induced L-[3H] noradrenalin (NA) release in brain slices, and this release was significantly lower in SAMP8 than in SAMR1 [43]. Damage to the central histaminergic neurons [44], synaptic dysfunction in the glutamatergic [45] and cholinergic systems [46,47] seem to be present in SAMP8. The SAMP8 retained a higher concentration of GM3 than SAMR1 throughout their life span [48]. The endogenous levels of the beta-subunit of NGF in these mice was already elevated in the thymus, adrenal gland, testes and hypophysis during the early period of life as compared to SAMR1 [49,50]. Neurotrophin-3 (NT-3) mRNA in the cortex was higher in SAMP8 than in SAMR1, whereas in the midbrain, hippocampus and forebrain, NT-3 expression levels were lower in SAMP8 than in SAMR1 during early development [51]. Brain glucose metabolism was also impaired, as indicated by reductions in 2-deoxyglucose uptake [52,53] and in hexokinase activity [52] in aged SAMP8. The binding of [3H]phorbol-12,13-dibutyrate to protein kinase C in both cytosol and membrane fractions in the hippocampus of aged SAMP8 was reduced [54].
Senile amyloidosis (SAMP1)
Senile amyloidosis in laboratory mice was detected in the SAMP1 first [55, 56], and then following studies revealed that this pathology could also be widely observed in other strains of laboratory mice [57]. Fine amyloid fibrils were observed to be deposited in the extracellular space with advancing age in all of the organs, except for the bone marrow and brain parenchyma. The intact apolipoprotein A-II in the high-density lipoprotein (HDL) particle deposits as amyloid fibrils without degradation [58,59]. The decreased serum concentration of apo A-II protein in aged SAMP1 [60] may be caused by a decrease in the level of its mRNA in the liver [61]. The serum half-life of apo A-II decreased significantly, and was less than the apo A-I half-life in SAMP1 at 6 and 12 months of age [62].
Three variants of the apo A-II protein are present in the inbred strains of laboratory mice [63]. The molecular variant of apo A-II correlated with susceptibility to senile amyloidosis [64,65]. All three types are observed in the SAM strains of mice [63,66]. Studies using the apo A-II congenic strain, R1.P1-Apoa2c, revealed that Apoa2c alleles caused severe senile amyloidosis that shortens the life span, but does not change the mortality rate doubling time [65.
Age-related decline in immune responsiveness (SAMP1, SAMP2, SAMP8)
The antibody-forming capacity to a T-independent antigen, DNP-Ficoll and natural killer cell activity in SAMP1 and SAMP2 showed a marked decline with aging. In contrast, cytotoxic activity was not impaired [67,68].These defective antibody responses to a T-dependent antigen in vitro resulted from impaired T-helper activity ('Th 2'-like) [69]. A cross mating study of SAMP1 (low responder) with MHC-identical B10.BR (high responder) strongly suggested that the impaired 'Th2'-like activity was under the control of at least two genes. One of these genes was closely linked to the albino coat-color (c) and Gpi-1 (glucose phosphate isomerase-1) loci but not to Hbb (hemoglobin b chain) on chromosome 7 [70].
SAMP8 mice also showed a more rapid decline in the lymphoproliferative response to concanavalin A, but cytotoxic T lymphocyte responses did not change with age. Natural killer cell activity was reduced with age in both SAMR1 and SAMP8, but no strain difference was observed [71].
Sera from SAMP1 mice contained a number of autoantibodies, including natural thymotoxic autoantibody, anti-nuclear antibodies and IgG anti-single-stranded and anti-double-stranded DNA antibodies, and anti collagen type-II antibody[72, 73]. Age-associated glomerular mesangial and capillary lesions, with granular IgG and C3 deposition were also frequently observed in old SAMP1 [72,73].
A possible cellular mechanism of senescence acceleration and age-associated disorders
Other age-associated pathologies of SAM strains are described elsewhere [2,4,74,].
Recently, geriatric (age-associated) disorders in humans have been classified into two categories: age-related disorders and age-dependent disorders [75]. The former category was thought to be "simply prevalent in the advanced years of life offered the opportunity for prevention, control, or treatment" and the latter category was thought to be an inevitable part of senescence as a direct consequence of physiological senescence". The age-associated pathological phenotypes observed in the SAMP strains contain many "age-dependent" disorders. This means age-dependent functional declination of parenchymal cells of the tissues occurs in accelerated and emphasized manner in SAMP and these functional changes results in the senile degenerative disease.
One possible mechanism, which can accelerate the senescence process and can cause and/or exacerbate these age-associated pathologies in SAMP at early calendrical time, is hyperoxidative stress. Several studies have already reported a high oxidative status in SAMP mice in vivo as compared with SAMR1 mice, and a higher susceptibility to oxidative stress in SAMP mice in vivo as compared with SAMR1 mice [76-87]. Especially in the brain, the content of lipid peroxide was shown to increase with age [79] and become slightly but significantly higher in 11- to 12-month-old SAMP8 males than in SAMR1 males [77]. The SOD activity in the SAMP8 mouse brains was higher than that in the ddY mouse brains at both 3 and 11 months of age [79]. Sato et al. [85], however, observed higher lipid peroxide and protein carbonyl contents transiently at 4 to 8 weeks of age in the cerebral cortex but not in other regions. In this period, the net generation of reactive oxygen species increased in cerebral cells, while glutamine synthetase, an enzyme highly sensitive to reactive oxygen, was decreased in activity [85]. The catalase activity decreased by 75% and the acyl-CoA oxidase, a microperoxisomal H2O2 -producing enzyme, increased 1.6 hold [83]. The cortical synaptosomal membranes from 10-month-old SAMP8 mice showed structural characteristics of free radical oxidative stress in terms of electron paramagnetic resonance spin labeling (EPR), protein carbonyl content and activity of glutamine synthetase. Furthermore, 2-week treatment of SAMP8 mice at this age with a free radical scavenger, N-tert-butyl-alpha-phenylnitrone, caused a return toward normal values of the relevant EPR parameters [84].
In addition to these observations, evidence showing that free radical oxidative stress is induced by age-associated mitochondrial dysfunction is accumulating. In the mitochondria isolated from 18-month-old SAMP8 male mice liver, efficiency of ATP synthesis was depressed and a dysfunctional energy transfer mechanism was detected [86]. The mitochondria from the brain homogenates of 2-month-old SAMP8 males demonstrated a higher redox state and a higher activity of respiration with lower respiration control ratio than the mitochondria from SAMR1 mice [87]. Accelerated increase in chromosome aberration of bone marrow cells in SAMP1 mice [88], in mutation rate of Hprt locus of splenic lymphocytes in SAMP1 mice [89] and the higher rate of mitochondrial DNA deletion in SAMP8 mice brain [90] might be explained as the adverse effect of this hyperoxidative status.
Caloric Restriction in SAM Mice
There have been many attempts to intervene the senescence acceleration and specifically in these age-associated pathologies: senile osteoporosis, senile amyloidosis and age-associated deficit in learning and memory. These attempts including caloric restriction, administration of nutrients, chemicals and traditional herbal medicines, show beneficial effects on the aging process of SAM mice. The details are described elsewhere [74,91].
Caloric restriction on SAM mice
We describe the effects of caloric restriction on the senescence of SAM mice as an example. Chronic caloric restriction markedly improved the senescence acceleration (Fig. 6) and senile amyloidosis of SAMP1.
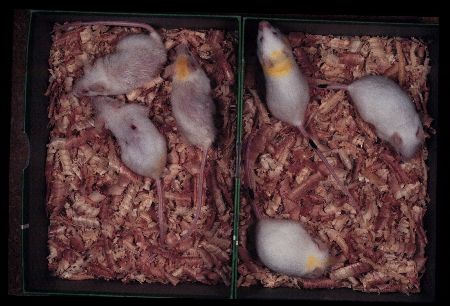 Fig. 6: SAMP1 females at 10 months of age fed ad libitum (left) and fed 40% restricted diet (right). Degree of senescence of restricted group is about a half of that observed in ad libitum fed group.
Fig. 6: SAMP1 females at 10 months of age fed ad libitum (left) and fed 40% restricted diet (right). Degree of senescence of restricted group is about a half of that observed in ad libitum fed group.
In a group fed 60% of control purified diet intake, the progress of senescence was significantly retarded and mean life span and 10th decile were prolonged. These improvement was not evident in SAMR1 groups. The severity of senile amyloid deposition was significantly less with 60% group than in the control group. Incidence of inflammatory changes and tumors were not affected [92]. The immune dysfunction associated with aging was improved by caloric restriction acceleration [93].
The SAM strains of mice have provided a unique model system to study the senescence in higher organisms and they would provide us clues to develop the applicable procedures to prevent or to control the age-associated disorders in humans.
References
1. Takeda, T., et al. new murine model of accelerated senescence. Mech. Ageing Dev. 1981; 17: 183-194.
2. Takeda,T et al.: Senescence-Accelerated Mouse(SAM): A novel murine model of accelerated senescence. J. Amer. Geriatr. Soc. 1991; 39: 911-919.
3. Takeda, T. et al.: Senescence-Accelerated Mouse (SAM). A novel murine model of aging. In Takeda, T. (ed.): The SAM Model of Senescence. Amsterdam: Excerpta Medica, 1994, pp.15-22.
4. Takeda T. et al.: Senescence-Accelerated Mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997; 32: 105-109.
5. Hosokawa M. et al.: Management and design of the maintenance of SAM mouse strains; an animal model for accelerated senescence and age associated disorders. Exp. Gerontol. 1997; 32: 111-116.
6. Hosokawa M. et al.: Grading score system: A method for evaluation of the degree of senescence in Senescence Accelerated Mouse (SAM). Mech. Ageing Dev. 1984; 26: 91-102.
7. Hosokawa, M.: Grading score system; a method of evaluation of the degree of senescence in Senescence-Accelerated Mouse (SAM).In Takeda, T. (ed.): The SAM Model of Senescence. Amsterdam: Excerpta Medica, 1994, pp.23-28.
8. Kitado H. et al.: Molecular genetic characterization of the Senescence-Accelerated Mouse (SAM) Strains, J. Gerontol. 1994; 49: B247-B254.
9. Takeda, T. et al.: Pathobiology of the Senescence-Accelerated Mouse (SAM). Exp. Gerontol. 1997; 32: 117-128.
10. Strehler, B.L. In Time, cells and Aging 2nd ed. New York, San Francisco, London: Academic Press, 1977.
11. Goldstein, S.: Human genetic disorders that feature premature onset and accelerated progression of biological aging. In Schneider Edward L. (ed.): The genetics of aging. New York: Plenum Press, 1978, pp. 171-224.
12. Zurcher, C., et al.: Aging Research. In Foster, H.L. et al. (eds.): The Mouse In Biomedical Research, vol. IV. New York, London, Paris, San Diego, San Francisco, Sao Paulo, Sydney, Tokyo, Toronto: Academic Press, 1982. pp.11-35.
13. Hosokawa, M. et al.: Accelerated aging of dermal fibroblast-like cells from senescence-accelerated mouse (SAM). 1. Acceleration of population aging in vitro. Mech. Ageing Dev. 1994; 74: 65-77.
14. Lecka-Czernik, B., et al. Cellular and molecular biomarkers indicate precocious in vitro senescence in fibroblasts from SAMP6 mice: evidence supporting a murine model of premature senescence and osteopenia. I. Gerontol. 1997; 52A: B331-336.
15. Fujisawa, H., et al.: Accelerated aging of dermal fibroblast-like cells from the senescence-accelerated mouse (SAM): Acceleration of changes in DNA ploidy associated with in vitro cellular aging. J. Gerontol. 1998; 53A; B11-17.
16. Matsushita, M. et al.: Age-related changes in bone mass in the Senescence-Accelerated Mouse(SAM) SAM-R/3 and SAMP/6 as new murine models for senile osteoporosis. Am. J. Pathol. 1986; 215: 276-283.
17. Suda, T., et al.: Osteoporotic bone changes in SAMP6 are due to a decrease in osteoblast progenitor cells. In Takeda Toshio (ed.): The SAM Model of Senescence. Amsterdam: Excerpta Medica, 1994, pp. 47-52.
18. Jilka, R. L., et al.: Linkage of decreased bone mass with impaired osteoblastogenesis in murine model of accelerated senescence. J. Clin. Invest. 1996; 97: 1732-1740.
19. Okamoto, Y., et al.: Femoral peak bone mass and osteoclast number in an animal model of age-related spontaneous osteopenia. Anat. Rec. 1995; 242: 21-28.
20. Tsuboyama, T., et al.: Cross-mating study on bone mass in the spontaneously osteoporotic mouse (SAM-P/6). Bone Miner. 1993; 23: 57-64.
21. Shimizu, H., et al.: Identification of peak bone mass QTL using a spontaneously osteoporotic mouse strain. Mammalian Genome, in press.
22. Miyamoto, M. et al.: Age -related changes in learning and memory in the Senescence-Accelerated Mouse (SAM). Physiol. and Behav. 1986; 38: 399-406.
23. Yagi, H., et al.: Age-related deterioration of ability of acquisition in memory and learning in senescence accelerated mouse: SAM-P/8 as an animal model of disturbances in recent memory. Brain Res. 1988; 474: 86-93.
24. Miyamoto M.: Characteristics of age-related behavioral changes in Senescence-Accelerated Mouse SAMP8 and SAMP10. Exp. Gerontol. 1997; 32: 139-148.
25. Shimada A. et al. : Age-related deterioration in conditional avoidance task in the SAM-P/10 mouse, an animal model of spontaneous brain atrophy. Brain Res. 1993; 608: 266-272.
26. Flood, J.E. and Morley, J.E.: Age-related changes in foot shock avoidance acquisition and retention in senescence accelerated mouse (SAM). Neurobiol. Aging 1993; 14: 153-157.
27. Ohta A., et al.: Behavioral characteristics of the SAM-P/8 strain in Sidman active avoidance task. Brain Res. 1989; 498: 195-198.
28. Miyamoto, M. et al.: Senescence-accelerated mouse (SAM): Age-related reduced anxiety-like behavior in the SAM-P/8 strain. Physiol. Behav. 1992; 51: 979-985.
29. Yagi, H., et al.: Spontaneous spongy degeneration of the brain stem in SAM-P/8 mice, a newly developed memory-deficient strain. J. Neuropathol. Exp. Neurol. 1989; 48: 577-590.
30. Akiyama, H., et al.: Periodic acid-Schiff(PAS)-positive, granular structures increase in the brain of senescence accelerated mouse (SAM). Acta Neuropathol. 1986; 72: 124-129. 31. Kawamata, T. et al.: Neuropathological studies on strains of Senescence-Accelerated Mice (SAM) with age-related deficits in learning and memory. Exp. Gerontol. 1997; 32: 161-170.
32. Amano, T. et al.: Increased expression of cathepsins E and D in reactive microglial cells associated with spongiform degeneration in the brain stem of Senescence-Accelerated Mouse. Exp. Neurol. 1995; 136: 171-182.
33. Nakamura, S., et al: Monoamine oxidase-B-positive granular structures in the hippocampus of aged senescence-accelerated mouse (SAMP8). Acta Neuropathol. 1995; 90: 626-632.
34. Takemura, M., et al.,: Beta/A4 protein like immunoreactive granular structures in the brain of Senescence-Accelerated Mouse(SAM). Am. J. Pathol. 1993; 142: 1887-1897.
35. Akiguchi, I., et al.: Age-related morphological changes in the brain of Senescence-Accelerated Mouse (SAMP8). In Takeda Toshio (ed.): The SAM Model of Senescence. Amsterdam: Excerpta Medica, 1994, pp. 67-72.
36. Shimada, A. et al.: Inbred SAM-P/10 as a mouse model of spontaneous, inherited brain atrophy. J. Neuropathol. Exp. Neurol. 1992; 51: 440-450.
37. Ohnishi, K. et al.,: Age-related decrease of nerve growth factor-like immunoreactivity in the basal forebrain of senescence-accelerated mice. Acta Neuropathol. 1995; 90: 11-16.
38. Ueno, M., et al.: Age-related changes in barrier function in mouse brain I. Accelerated age-related increase of brain transfer of serum albumin in accelerated senescence prone SAM-P/8 mice with deficits in learning and memory. Arch. Gerontol. Geriatr. 1993; 16: 233-248.
39. Ueno, M., et al.: Immunocytochemical evaluation of the blood-brain barrier to endogenous albumin in the olfactory bulb and pons of senescence accelerated mice (SAM). Histochem. Cell Biol. 1996; 105: 203-212.
40. Vorbrodt, A.W., et al.: A quantitative immunocytochemical study of blood-brain barrier to endogenous albumin in cerebral cortex and hippocampus of senescence-accelerated mice (SAM). Folia Histochem. et Cytobiol. 1995; 33: 229-237.
41. Ueno, M. et al.: Age-related changes in the brain transfer of blood-borne horseradish peroxidase in the hippocampus of Senescence-Accelerated Mouse (SAM), Acta Neuropathol. 1997; 93: 233-240.
42. Kitamura, Y., et al.: Ligand-binding characteristics of [3H] QNB, [3H] Prazosin, [3H] rauwolscine, [3H] TCP and [3H] nitrendipine to cerebral cortical and hippocampal membranes of senescence accelerated mouse. Neurosci. Lett. 1989; 106: 334-338.
43. Zhao, X-H, and Nomura, Y.: Age-related changes in uptake and release on L-[3H] noradrenaline in brain slices of Senescence Accelerated Mouse. Int. J. Devl. Neuroscience. 1990; 8: 267-272.
44. Meguro, K., et al.: Neurochemical studies on central histaminergic neuron system of senescence accelerated mouse. Biogenic Amines 1992; 8: 299-307.
45. Kitamura, Y., et al: Age-related changes in transmitter glutamate and NMDA receptor/channels in the brain of senescence-accelerated mouse. Neurosci. Letter 1992; 137: 169-162.
46. Ikegami, S., et al.: Age-related changes in radial-arm maze learning and basal forebrain cholinergic systems in senescence accelerated mice (SAM). Behav. Brain Res. 1992; 51: 15-22.
47. Zhao, X-H. et al.: Age-related changes in NMDA-induced [3H] acetylcholine release from brain slices of Senescence-Accelerated Mouse. Int. J. Devl. Neuroscience. 1992; 10: 121-129.
48. Ohsawa, T. and Shumiya, S.: Age-related alteration of brain gangliosides in Senescence-Accelerated Mouse (SAM)-P/8. Mech. Ageing Devl. 1991; 59: 263-274.
49. Katoh-Semba R., et al.: Elevated concentrations of b-nerve growth factor in selected tissues from Senescence Accelerated Mice (SAM-P/8). Mech. Ageing Devl. 1991; 59: 163-175.
50. Katoh-Semba R., et al.: An acceleration of age-related increases in levels of the beta-subunit of nerve growth factor in selected tissues from Senescence-Accelerated Mice (SAM-P/8). J. Mol. Neurosci.1993; 4: 107-115.
51. Kaisho, Y., et al.: Expression of neurotrophin genes in the brain of senescence-accelerated mouse (SAM). Brain Res. 1994; 647: 139-144.
52. Kurokawa, T. et al.: Evidence that glucose metabolism is decreased in the cerebrum of aged female senescence-accelerated mouse; possible involvement of a low hexokinase activity. Neurosci. Letter 1996; 214: 45-48.
53. Ohta, H., et al.: Relationship of impaired brain glucose metabolism to learning deficit in senescence-accelerated mouse. Neurosci. Letter, 1996; 217: 37-40.
54. Nomura, Y., et al.: Alterations in acetylcholine, NMDA, benzodiazepine receptors and protein kinase C in the brain of senescence-accelerated mouse: an animal model useful for studies on cognitive enhancers. Bihav. Brain Res. 1997; 83: 51-53.
55. Matsumura, A., et al.: A novel amyloid fibril protein isolated from senescence-accelerated mice. Lab. Invest. 1982;, 47: 270-275.
56. Takeshita, S., et al.: Spontaneous age-associated amyloidosis in Senescence-Accelerated Mouse(SAM). Mech. Ageing Devl. 1982; 20: 13-23.
57. Higuchi, K., et al.: Mouse senile amyloidosis ASSAM amyloidosis in mice presents universally as a systemic age-associated amyloidosis. Virchows Arch. B Cell Pathol.1991; 60: 231-239.
58. Higuchi, K., et al.: Purification and characterization of a senile amyloid-related antigenic substance (apoSASSAM) from mouse serum. J. Biol. Chem. 1986; 261: 12834-12840.
59. Higuchi, K., et al.: The single proline-glutamine substitution at position 5 enhances the potency of amyloid fibril formation of murine apo A-II. FEBS Lett. 1986; 207: 23-27.
60. Higuchi, K., et al.: Age-related changes of serum apoprotein SASSAM, apoprotein A-I and low-density lipoprotein levels in Senescence Accelerated Mouse(SAM). Mech. Ageing Devl. 1984; 26: 311-326.
61. Kitagawa, K., et al.: Age-associated decreases in the Messenger Ribonucleic Acid level and the rate of synthesis of Apolipoprotein A-II in murine senile amyloidosis. Lab. Invest. 1994; 70: 565-571.
62. Naiki, H., et al.: Metabolism of senile amyloid precursor and amyloidogenesis. Age-related acceleration of apolipoprotein A-II clearance in the Senescence Accelerated Mouse. Am. J. Pathol. 1988; 130: 579-587.
63. Higuchi, K., et al.: Polymorphism of apolipoprotein A-II(apoA-II) among inbred strains of mice. Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Bioch. J. 1991; 279: 427-433.
64. Higuchi, K., et al.: Apolipoprotein A-II gene and development of amyloidosis and senescence in a congenic strain of mice carrying amyloidogenic ApoA-II. Lab. Invest. 1995; 72: 75-82.
65. Higuchi, K., et al.: Accelerated senile amyloidosis induced by amyloidogenic ApoA-II gene shortens the life span of mice but does not accelerate the rate of senescence, J. Gerontol. 1996; 51A: B295-302.
66. Higuchi, K.: Genetic characterization of Senescence-Accelerated Mouse (SAM). Exp. Gerontol. 1997; 32: 129-138.
67. Hosokawa, T., et al.: Immune responses in newly developed short-lived SAM mice I. Age-associated early decline in immune activities of cultured spleen cells. Immunology 1987; 62: 419-423.
68. Hosokawa, T., et al.: Immune responses in newly developed short-lived SAM mice II. Selectively impaired T-helper cell activity in in vitro antibody response. Immunology 1987; 62: 425-429.
69. Hanada K., et al. : Immune responses in newly developed short-lived SAM mice III. Genetic control of defective helper T-cell activity in in vitro primary antibody response. Immunology 1989; 68: 540-546.
70. Hanada K., et al.: Immune responses in newly developed short-lived SAM mice IV. Chromosomal location of a gene controlling defective helper T-cell activity. Immunology 1991; 74: 160-164.
71. Powers, D.C., et al.: Age-related changes in lymphoproliferation, cytotoxic T lymphocyte responses, and natural killer activity of Senescence-Accelerated Mouse (SAM)-P/8 and SAM-R/1 substrains. Aging: Immunol. Infec. Dis. 1995; 6: 43-52.
72. Yoshioka, H., et al.: Autoimmune abnormalities in a murine model of accelerated senescence. Clin. Exp. Immunol. 1989; 75: 129-135.
73. Yoshioka, H., et al.: Spontaneous development of anti-collagen type II antibodies with NTA, and anti-DNA antibodies in Senescence-Accelerated Mice. Autoimmunity 1993; 14: 215-220.
74. The SAM Model of Senescence. Takeda Toshio (ed.): Amsterdam: Excerpta Medica, 1994.
75. Cotran, R.S.: Diseases of Aging. In Cotran, R.S., et al. (eds.): Robbins Pathologic Basis of Disease, 4th ed. Philadelphia, London, Toronto, Montreal, Sydney, Tokyo: W.B. Saunders Company, 1989. pp. 543-551.
76. Komura, S. et al.: Lipid peroxide levels in the skin of the Senescence-Accelerated Mouse. J. Clin. Biochem. Nutr. 1988; 5: 255-260.
77. Nomura, Y., et al.: Biochemical changes related to aging in the Senescence-Accelerated Mouse. Exp. Gerontol. 1989; 24: 49-55.
78. Teramoto, S., et al.: Age-related changes in GSH content of eyes in mice-A comparison of Senescence-Accelerated Mouse (SAM) and C57BL/j mice. Comp Biochem. Physiol. 1992; 102A: 693-696.
79. Liu, J. and Mori, A.: Age- associated changes in superoxide dismutase activity, thiobarbituric acid reactivity and reduced glutathione level in the brain and liver in senescence accelerated mice (SAM): a comparison with ddy mice. Mech. Ageing Dev. 1993; 71: 23-30.
80. Uejima, Y., et al.: Age changes in visceral content of glutathione in the Senescence Accelerated Mouse (SAM). Mech Ageing Dev. 1993; 67: 129-139.
81. He, P., et al.: Age-related changes in glutathione concentration, glutathione peroxidase, glutathione S transferase, and superoxide dismutase activities in Senescence Accelerated Mice. Biosci. Biotech. Biochem. 1994; 58: 1037-1040.
82. Park, J-W., et al.: Oxidative status in Senescence-Accelerated Mice. J. Gerontol. 1996; 51A: B337-345.
83. Sato, E. et al.: Early appearance of abnormality of microperoxisomal enzymes in the cerebral cortex of senescence-accelerated mouse. Mech. Ageing Dev. 1996; 92: 175-184.
84. Butterfield, A.D., et al.: Free radical oxidation of brain proteins in Accelerated Senescence and its modulation by N-tert-butyl-alpha-phenylnitrone (PBN). Poc. Nat. Acad. Sci. U.S.A. 1997; 94: 675-678.
85. Nakahara, H., et al.: Mitochondrial dysfunction in the senescence accelerated mouse (SAM). Free Rad. Biol. Med. 1998; 24: 85-92.
86. Sato, E., et al.: Early and transient increase in oxidative stress in the cerebral cortex of senescence-accelerated mouse. Mech. Ageing Dev. 1996; 86: 105-114.
87. Nishikawa, T., et al.: An early stage mechanism of the age-associated mitochondrial dysfunction in the rein of SAMP8 mice; an age-associated neurodegeneration animal model. Neurosci. Letters. 1998; 254: 69-72.
88. Nisitani, S., et al.: Acceleration of chromosome aberrations in senescence-accelerated strains of mice. Mut. Res. 1990; 237: 221-228.
89. Odagiri, Y., et al.: Accelerated accumulation of somatic mutations in the senescence-accelerated mouse. Nature Genetics 1998: 19: 117-118.
90. Fujibayashi, Y., et al.: Increased mitochondrial DNA deletion in the brain of SAMP8, a mouse model for spontaneous oxidative stress brain. Neurosci. Letters 1998; in press.
91. Hosokawa, M., et al.: Interventions of senescence in SAM mice. J. Anti-aging Med. 1998; 1:27-37.
92. Kohno, A., et al.: Chronic food restriction modulates the advance of senescence in the Senescence Accelerated Mouse(SAM). J. Nutr. 1985; 115: 1259-1266.
93. Umezawa, M., et al.: Effects of dietary restriction on age-related immune dysfunction in the Senescence Accelerated Mouse (SAM). J. Nutr. 1990; 120: 1393-1400.
| Discussion Board | Previous Page | Your Symposium |