Invited Symposium: Molecular Mechanisms of Ageing
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Overview
We can easily distinguish an old person from a young person. However, the aging process varies greatly with the individual. Our maximum lifespan is limited and nobody has ever lived over 150 years. Thus aging is a variable but tightly programmed biological phenomenon. We know very little why and how we age. Elucidation of the mechanism of aging and deceleration of aging is one of the final frontiers of biomedical science. Although recent advances in molecular biology are giving us many epoch-making findings to solve the exciting mystery, the "Molecular Mechanism of Aging," we are far from the establishment of a general concept, which explains the aging process. Now we need to collect, accumulate and analyze as much information as possible by the researchers in various areas of science.
Thus, I organized this symposium "Molecular Mechanism of Aging," and selected ten invited speakers who are studying aging using various methods and animal species. Dr. Takagi is trying to create a new concept of cellular lifespan using Paramedium. Dr. Ishii has used nematode to analyze the etiology of oxidative damage in aging. Dr. Hosokawa is analyzing aging and age-associated disorders using a mouse model of accelerated senescence (SAM: senescence accelerated mouse). Dr. Noda has been using molecular biological techniques to elucidate the mechanism of senescence growth arrest in human cultured cells. Dr. Toda is using a proteome technique to analyze age-associated changes in protein expression. Dr. Goto has biochemically studied the modificationof proteins during aging. Dr. Chung is investigating energetically the repair system of oxidized DNA. Dr. Naiki has been investigating the mechanism of Abeta amyloid fibril formation using an in vitro system. Dr. Shimada has studied morphologically the cultured Purukinje cell. Finally, Dr. Johnson a opinion leader of the aging science, will summarize the important roles of stress response in aging and longevity.
I have been using mouse senile amyloidosis as an unique model of age related degeneration of functional protein. Recently we established a concept of �fibril conformation dependent polymerization model� for amyloid fibril formation and found the transmission of amyloid fibril through the digestive tract. I would like to present our recent studies.
Mouse senile amyloidosis
Innocuous and soluble proteins polymerize to insoluble amyloid fibrils in several serious diseases including Alzheimer's disease, transmissible prion disease, and familial amyloid diseases. Although the diverse proteins that can polymerize into amyloid fibrils have unrelated sequences, they can all form fibrils with a similar ultrastructural appearance and a similar core structure consisting of beta-pleated-sheets (1-3). Nucleation-dependent polymerization is postulated to be a model which explains well the kinetics of fibrillization of amyloid proteins in AD and prion disease(4-6). This model consists of two phases, i.e., nucleation and extension phases. Nucleus formation requires a series of association steps of monomers, that are thermodynamically unfavorable, representing the rate-limiting step in amyloid fibril formation. Once the nucleus has been formed, further addition of monomers becomes thermodynamically favorable, resulting in rapid extension of amyloid fibrils. Addition of nucleus (amyloid fibrils) in vitro into the solution of amyloid protein monomers dramatically hastens the fibrillization (7-9)
. Apolipoprotein A-II (apoA-II), deposits as an amyloid fibril (AApoAII) in murine senile amyloidosis (10-12). Using mouse senile systemic amyloidosis, we examined whether the nucleation-dependent polymerization occurs in vitro as a fundamental model to explain the mechanisms of various amyloidosis.
1) We induced severe systemic amyloid deposition in young R1.P1-Apoa2c mice which have amyloidogenic type C apoA-II by a single intravenous injection of a very small amount of the native mouse senile amyloid fibrils (AApoAII).
2) The importance of the fibril conformation for the injected amyloid fibrils to act as seeds in vivo was studied by the injection of denatured AApoAII, native apoA-II in high density lipoprotein (HDL) and denatured apoA-II monomer which have the same primary structure but without a fibril conformation.
3) AApoAII fibrils were injected intravenously into three kinds of congenic strains of mice each of which has a different type of apoA-II gene.
4) Transmissibility of the amyloid through the digestive tract was examined by giving water containing AApoAII fibrils.
1) Acceleration of amyloidosis by AApoAII amyloid fibrils
Two-month-old male R1.P1-Apoa2c mice were injected intravenously with AApoAII fibrils isolated and purified from the liver of old R1.P1-Apoa2c congenic mice. The control mice were injected with distilled water (DW). Slight amyloid deposition in the submucosal tissues of the tongue, small intestine and the stomach was detected in all AApoAII-injected mice (7/7) as early as one month after injection and the intensity of the amyloid deposition (both the number of amyloid deposited tissues and deposited area in each tissue) increased rapidly there-after (Fig.1).
At three months after the injection there was severe amyloid deposition throughout the whole body. The systemic severe distribution of AApoAII amyloid deposits in mice at four months after injection was the same as that occurring in spontaneous senile amyloidosis in old (over 15 months) R1.P1-Apoa2cmice (13). On the other hand, amyloid deposition began later and its intensity increased much more slowly in mice injected with DW. Thus, a single injection of a small amount of AApoAII amyloid fibrils accelerated the amyloid deposition markedly without a lag time.
2) Importance of fibril conformation for acceleration of amyloidosis. To study the importance of fibril-conformation in the acceleration of amyloid deposition, we injected mice with four proteins, native AApoAII fibrils, guanidine denatured amyloid fibrils (GDAM), serum HDL containing native type C apoA-II and urea-denatured type C apoA-II. Although the native AApoAII fibrils induced amyloid deposition in mice, GDAM, native and denatured serum apoA-II protein did not. 3) The role of the primary structure of endogenous apoA-II protein. Three congenic strains of mice each having a different type of apoA-II gene on the same genome of the SAMR1 strain were injected intravenously with AApoAII fibrils (0.1 mg). No amyloid deposition was observed three months after the injection in SAMR1 mice having a type B apoA-II gene. Amyloid deposition was observed in all mice having the type B and type C apoA-II gene heterozygously at 3 months after the AApoAII injection. However, degree of amyloid deposits (AI) was significantly lower in heterozygous mice. 4) Transmission of the AApoAII amyloid through the digestive tract Amyloid deposits were detected in the small intestine of all mice 3 months after drink of 2.5 mg of AApoAII fibrils suspended in water. Amyloid deposits were observed in the lamina propria and submucosa of the small intestine, collecting tubules in the papillae of the kidneys and squamous-glandular junction of the stomach. These findings suggested that the nucleation-dependent polymerization found in vitro (14) also occurs in vitro and that the fibril conformation is required for the injected amyloid fibrils to act as seeds in vivo. Amyloid deposition was accelerated in mice drinking water containing AApoAII fibrils, suggesting the oral transmission of systemic amyloidosis. The acceleration of amyloid deposition may be a primary event in diseases with amyloid deposition, i.e., Alzheimer�s disease, Creutzfeldt-Jakob disease, Bovine spongiform encephalopathy, familial amyloid polyneuropathy, AA and human senile systemic amyloidosis. Thus, endogenous and exogenous (environmental) factors that affect the rate of amyloid formation may play a significant role in the pathogenesis of these amyloid-related diseases. In mouse senile AApoAII amyloidosis, calorie restriction, rearing of mice in specific pathogen-free conditions, treatment with immuno-suppresive agents and bone marrow transplantation suppressed the amyloid deposition. The present mouse model should provide a valuable system to study the general mechanism for fibrillization underling the amyloid-related diseases.
REFERENCES
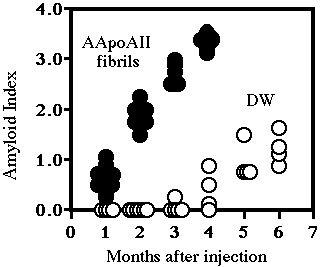 Fig. 1: Purified AApoAII fibrils were suspended in DW at the concentration of 1.0 mg/ml and sonicated thoroughly. Then, 0.1 ml of this suspension of AApoAII fibrils (closed circles) and 0.1 ml DW (open circles) were injected intravenously into the tail of 2-month-old male mice. Twenty-four mice were injected with AApoAII fibrils and killed at 1, 2, 3 and 4 months after the injection. Twenty-nine mice were injected with DW and killed at 1, 2, 3, 4, 5 and 6 months after the injection. Amyloid deposition in each tissue was detected in Congo red stained sections and the intensity of the amyloid deposition was determined using the Amyloid Index (AI) as a parameter..
Fig. 1: Purified AApoAII fibrils were suspended in DW at the concentration of 1.0 mg/ml and sonicated thoroughly. Then, 0.1 ml of this suspension of AApoAII fibrils (closed circles) and 0.1 ml DW (open circles) were injected intravenously into the tail of 2-month-old male mice. Twenty-four mice were injected with AApoAII fibrils and killed at 1, 2, 3 and 4 months after the injection. Twenty-nine mice were injected with DW and killed at 1, 2, 3, 4, 5 and 6 months after the injection. Amyloid deposition in each tissue was detected in Congo red stained sections and the intensity of the amyloid deposition was determined using the Amyloid Index (AI) as a parameter..
| Discussion Board | Previous Page | Your Symposium |