Invited Symposium: On/Off Switches for Nitric Oxide Synthases
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Regulation of eNOS expression by Statins
Endothelial-derived nitric oxide (NO) is an important mediator of vascular function. through its ability to inhibit vasoconstriction, platelet aggregation, smooth muscle cell proliferation and leucocyte adhesion. Clinical studies indicate that cholesterol-lowering agents, HMG-CoA reductase inhibitors (statins), improve endothelial function in hypercholesterolemic and atherosclerotic individuals and suggest that statins may upregulate or enhance endothelial-derived NO. Since hypoxia causes endothelial dysfunction, in part, by decreasing endothelial NO synthase (eNOS) expression and activity, we sought to determine whether statins can prevent hypoxia-mediated downregulation of eNOS.
Accordingly, human endothelial cells were exposed to hypoxia (3% O2) in the presence of statins, simvastatin (Sim) and lovastatin. Exposure of endothelial cells to hypoxia decreased eNOS mRNA levels in a time-dependent manner resulting in a 9-fold reduction after 48 h (Fig. 1 A). In a concentration-dependent manner simvastatin not only prevented the downregulation of eNOS expression by hypoxia (Fig. 1 B) but also caused a direct increase in eNOS expression (Fig. 1 C). Simvastatin-induced changes in ecNOS expression correlated with changes in endothelial NO production and were reversed by treatment with L-mevalonate but not cholesterol. Nuclear run-on studies showed that simvastatin had no effect on ecNOS gene transcription. Instead, Actinomycin D studies revealed that under hypoxic condition, simvastatin increased ecNOS mRNA half-life from 13 to 38 h. These results indicate that statins regulate ecNOS function and expression through changes in ecNOS mRNA stability. Figure 1 (adapted from Ref. 1 and 2):
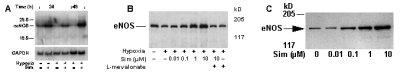 Figure 1
Figure 1
Rho negatively regulates eNOS expression
Inhibition of mevalonate synthesis by statins not only blocks the formation of cholesterol but also of isoprenoids, such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP), who are important for the posttranslational modification of signaling proteins (Fig. 2). The upregulation of eNOS expression by statins was mediated via the inhibition of geranylgeraniol, but not farnesiol. Co-treatment with L-mevalonate or GGPP, but not FPP or LDL-cholestgerol, reversed the statin effects. Since Rho GTPases are predominant proteins which undergo geranylgeranyl modification, we investigated whether Rho negatively-mediates statins� effects on eNOS expression. Immunoblot analyses and [35S]-GTPgS-binding assays revealed that statins inhibited Rho membrane translocation and GTP-binding activity. Inhibition of Rho by Clostridium botulinum C3 transferase or by overexpression of a dominant-negative N19RhoA mutant increased eNOS expression (Fig. 2). In contrast, direct activation of Rho by Escherichia coli cytotoxic necrotizing factor (CNF)-1 decreased eNOS expression. These findings indicate that Rho negatively regulates eNOS expression and that statins upregulate eNOS expression by blocking Rho geranylgeranylation which is necessary for its membrane-associated activity.
Figure 2 (adapted from Ref. 3):
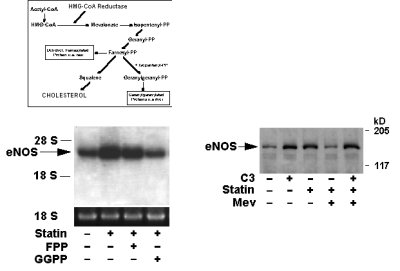 Figure 2
Figure 2
Upregulation of eNOS by Statins in vivo
Ischemic stroke is the third leading cause of death in the United States. Its treatment is limited to prophylactic agents which block the coagulation cascade. Since endothelial NO is an important determinant of cerebral blood flow and eNOS-deficient mice have enlarged cerebral infarcts, we hypothesized that statins through its effects on eNOS may be a useful trearment strategy for ischemic strokes. Indeed, we found that statins protect against cerebral injury in mice subjected to middle cerebral artery occlusion.. Prophylactic treatment with Sim augmented cerebral blood flow, reduced cerebral infarct size, and improved neurological function in normocholesterolemic mice (Fig. 3).
Figure 3 (adapted from Ref. 4) :
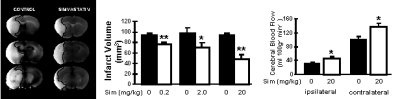 Figure 3
Figure 3
In addition, Sim increased eNOS expression (RT-PCR, Fig. 4 A) and activity (arginine assay, Fig. 4 B) in the murine aorta. Figure 4 (adapted from Ref. 4):
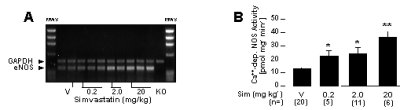 Figure 4
Figure 4
The upregulation of eNOS by HMG-CoA reductase inhibitors was not associated with changes in serum cholesterol levels, but was reversed by co-treatment with L-mevalonate. The blood flow and neuroprotective effects of HMG-CoA reductase inhibitors are completely absent in eNOS-deficient mice indicating that enhanced eNOS activity by HMG-CoA reductase inhibitors is the predominant if not the only mechanism by which these agents protect against cerebral injury. Our results suggest that HMG-CoA reductase inhibitors provide a novel prophylactic treatment strategy for increasing blood flow and reducing brain injury during cerebral ischemia.
References
1. Laufs U, La Fata V, and Liao JK. (1997) Inhibition of 3-hydroxy-3-methylglutaryl (HMG) CoA reductase blocks hypoxia-mediated downregulation of endothelial nitric oxide synthase. J. Biol. Chem. 272(50): 31730-35.
2. Laufs U, La Fata V, Plutzky J, and Liao JK. (1998) Upregulation of endothelial nitric oxide synthase by HMG-CoA reductase inhibitors. Circulation 97: 1129-35
3. Laufs U, and Liao JK. (1998) Post-Transcriptional Regulation of Endothelial Nitric Oxide Synthase mRNA Stability by Rho GTPase. J. Biol. Chem. (in press)
4. Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, and Liao JK. (1998) Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA:; 95(15): 8880-8885
| Discussion Board | Previous Page | Your Symposium |