Invited Symposium: Angiotensin Receptors
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Results
1. Conservation patterns in the sequences of GPCRs
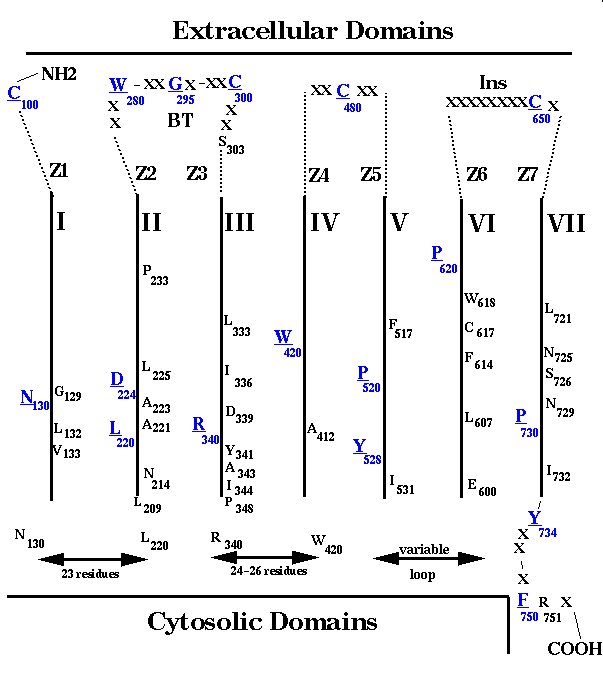
Fig. 1 shows the conserved residues (>50%) identified by a multiple alignment of about 1400 GPCRs. The numbering system is based on the conserved residue positions which were found in the transmembrane segments of rhodopsin-like receptors [1]. The limits for the transmembrane regions, which were based on conservation patterns obtained from multiple sequence alignment, are similar to those described by Baldwin et al. [25]. The very conserved positions are mostly found in the transmembrane regions but many are also observed at the adjacent loops. In the external side of receptors, these patterns suggest the existence of nine regions which were named Z1-Z7, BT and Ins in Fig. 1.
 Click to enlarge
Fig. 1: Conserved residues in a consensual sequence obtained from multiple sequence alignment of about 1400 rhodopsin-like GPCRs. The domains in the extracellular sequences Z1-Z7, BT, Ins; in the transmembrane regions I-VII; and in the cytosolic loop sequences are described in the text. Residues in blue and underlined are those found most conserved in the different domains and were used for numbering the sequences in these regions: Cys100 in region Z1; Asn130 in the region comprising helix I and the N-terminal half of loop I-II; Leu220 and Asp224 in the region comprising the C -terminal half of loop I-II and helix II; Trp280 in region Z2; Gly295 in region BT; Cys300 in region Z3; Arg340 in the region comprising helix III and the N-terminal half of loop III-IV; Trp 420 in helix IV; Cys480 in the region comprising the N-terminal (Z4) and C-terminal (Z5) portions of loop IV-V; Pro520 and Tyr528 in the region comprising helix V and the N-terminal portion of loop V-VI; Pro620 in the region comprising helix VI and the N-terminal portion of loop VI-VII (Z6); Cys650 in the region Ins; Pro730 in the region comprising the C-terminal portion of loop VI-VII (Z7) and helix VII; Tyr734 in the proximal portion of the C-terminal tail; Phe750 in the distal portion of the of C-terminal tail. The region Z1 is only found in the receptors for AngII, bradykinin, endothelin, chemokines and neuropeptides which contain the Ins region at the loop VI-VII and the supposed disulphide bridge Cys100-Cys650.
Click to enlarge
Fig. 1: Conserved residues in a consensual sequence obtained from multiple sequence alignment of about 1400 rhodopsin-like GPCRs. The domains in the extracellular sequences Z1-Z7, BT, Ins; in the transmembrane regions I-VII; and in the cytosolic loop sequences are described in the text. Residues in blue and underlined are those found most conserved in the different domains and were used for numbering the sequences in these regions: Cys100 in region Z1; Asn130 in the region comprising helix I and the N-terminal half of loop I-II; Leu220 and Asp224 in the region comprising the C -terminal half of loop I-II and helix II; Trp280 in region Z2; Gly295 in region BT; Cys300 in region Z3; Arg340 in the region comprising helix III and the N-terminal half of loop III-IV; Trp 420 in helix IV; Cys480 in the region comprising the N-terminal (Z4) and C-terminal (Z5) portions of loop IV-V; Pro520 and Tyr528 in the region comprising helix V and the N-terminal portion of loop V-VI; Pro620 in the region comprising helix VI and the N-terminal portion of loop VI-VII (Z6); Cys650 in the region Ins; Pro730 in the region comprising the C-terminal portion of loop VI-VII (Z7) and helix VII; Tyr734 in the proximal portion of the C-terminal tail; Phe750 in the distal portion of the of C-terminal tail. The region Z1 is only found in the receptors for AngII, bradykinin, endothelin, chemokines and neuropeptides which contain the Ins region at the loop VI-VII and the supposed disulphide bridge Cys100-Cys650.
Region Z1 can only be identified in the sequences of AngII II, bradykinin, endothelin, chemokine and neuropeptide receptors between the conserved Cys100 and the beginning of helix I. Regions Z2, Z3 and BT are in loop II-III. The number of residues in this loop is variable but multiple alignment shows high conservation for three residue positions (Trp280, Gly295 and Cys300). Region Z2 is the segment between helix II and Trp280. Region BT is between Trp280 and Cys300 and supposedly forms a beta-turn in about 65% of the GPCRs. A fixed sequence length is observed between Cys300 and Arg340 at the end of helix III (24 residues for the large majority of receptors). However, as residue insertions in the newly-cloned worm GPCR sequences have recently been observed after the conserved Cys300 [1], a gap was introduced at this position so that the numbering of the helix III sequence was exclusively based on Arg340. As a consequence, a region Z3 can be defined between this gap and Cys300 (Fig. 1). Regions Z4 and Z5 are respectively located before and after loop IV-V�s Cys480 which makes a disulphide bridge with Cys300. Regions Z6 and Z7 are located before and after an insertion of about eight residues (Ins) found in the middle of loop VI-VII in the same peptide receptors referred to above as containing the Cys100. Interestingly, this insertion, in all these receptors, contains the Cys650 which has been supposed to make a second extracellular disulphide bond with Cys100 [28].
Regarding the cytosolic side of GPCRs, some conservation patterns can be considered. For loop I-II, a conservation of sequence length (23 residues), but not of residues, is usually observed between Asn130 and Leu220 (Fig. 1). However, insertions have been observed in this region, as in bradykinin B1 receptors [29]. High conservation of residues is observed in a segment of loop III-IV adjacent to helix III (after Arg340) for all GPCRs, a region thought to be crucial for activating the G protein complex [30].
No appreciable levels of conservation are observed in loop V-VI of GPCRs, but remarkable conservation patterns are present at the distal portion of helix VII (Fig. 1). Besides the conserved P730, a Tyr residue occupies position 733 in 97% of GPCRs or position 734 in the remaining receptors. Therefore, a gap was added at position 733, and Tyr734 was considered the first residue of the receptor's C-terminal tail. After residue 734 the sequences are variable but become conserved again at the level of F750 and R751 (Fig. 1). This sequence profile supports the existence of a loop in the transition between helix VII and the C-terminal domain of GPCRs, rather than the prolongation of the a-helix. However, an a-helix was assumed after the FR doublet in the AT1 receptor model as a crucial motif for coupling to G-protein.
2. Molecular modeling of the AngII/AT1 receptor complex
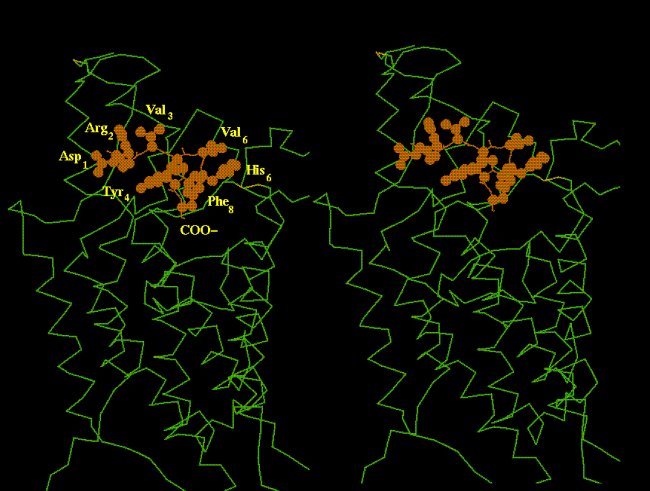
A functionally active conformation of AngII was determined by Nikiforovitch et al. [26] which consists of a beta-turn at the level of residues Val(Ile)5 and His6, so that the Tyr4 phenolic ring and the C-terminal carboxylate are pointing towards an opposite direction relative to the side-chains of Ile3, Val(Ile)5, His6 and Phe8 (Fig. 2).
 Click to enlarge
Fig. 2. Binding mode of AngII molecule relative to the seven-helix bundle of the AT1 receptor. Note that the side-chains of agonist residues Val3, Val5, His6 and Phe8 are pointing outwards while the C-carboxylate and the Tyr4 side chain are inserted into the transmembrane space. Peptide conformation was determined by Nikoforovitch et al. [26].
Click to enlarge
Fig. 2. Binding mode of AngII molecule relative to the seven-helix bundle of the AT1 receptor. Note that the side-chains of agonist residues Val3, Val5, His6 and Phe8 are pointing outwards while the C-carboxylate and the Tyr4 side chain are inserted into the transmembrane space. Peptide conformation was determined by Nikoforovitch et al. [26].
A preliminary requirement arisen from mutagenesis studies is that the AngII molecule should bind simultaneously to the receptor side-chains of Lys199(516) and Asp278(709) and Asp281(712), through the C-terminal carboxylate and Arg2 side-chain, respectively (numbers in parentheses refer to the normalized numbering system). To satisfy this requirement, the AngII molecule, with the conformation described above, could only be docked to the AT1 receptor as shown in Fig. 2. In this mode, the agonist's constrained side-chains of Val3, Val(Ile)5, His6 and Phe8 point outwards while Tyr4's side-chain and the C-terminal carboxylate are inserted in the receptor transmembrane pocket.
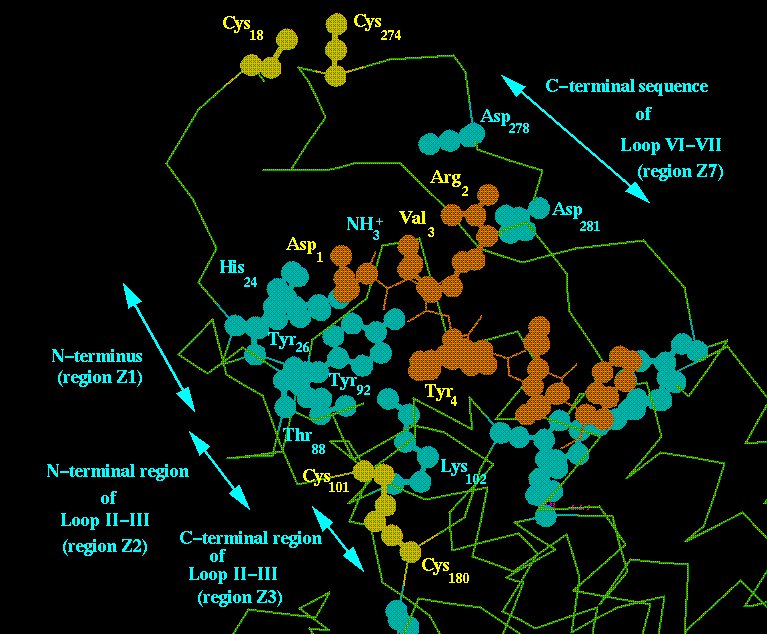
In the model of the AngII/AT1 receptor complex, the ligand's N-terminal half is placed at a locus surrounded by the receptor's regions Z1 and Z7 (Fig. 3).
 Click to enlarge
Fig. 3. AT1 receptor locus supposed to accommodate the N-terminal region of AngII molecule. The agonist molecule is drawn in brown and the receptor groups suggested by mutagenesis studies to be implicated in binding are shown in light blue. Disulphide bridges are shown in yellow. Note the well-discriminated distribution of the residues involved in binding in regions Z1, Z2, Z3, Z4 and Z7.
Click to enlarge
Fig. 3. AT1 receptor locus supposed to accommodate the N-terminal region of AngII molecule. The agonist molecule is drawn in brown and the receptor groups suggested by mutagenesis studies to be implicated in binding are shown in light blue. Disulphide bridges are shown in yellow. Note the well-discriminated distribution of the residues involved in binding in regions Z1, Z2, Z3, Z4 and Z7.
Besides the side-chains of Asp278(709) and Asp281(712), in region Z7, which supposedly interact with AngII's Arg2, other residues are found at this locus such as His24(106), Tyr26(107) and Ile27(108) (region Z1) which might bind the peptide's Asp1 side-chain and N-terminal amino group. A disulphide bond between Cys18(100) and Cys274(650) might contribute to stabilize this AngII-N-terminal binding locus. Only the initial and final portions of loop II-III (corresponding to regions Z2 and Z3) and region Z5 (Fig. 1) contain ligand-binding residues [4]. The intermediary region containing Gly295 (region BT in Fig. 1), is conserved for about 65% of GPCRs, including AT1, and supposedly makes a beta-turn probably involved in a general receptor function. Fig. 3 shows the possibility that AT1 residues Thr88(274), Tyr92(278) and Lys102(301), present in regions Z2 and Z3 of loop II-III, could interact with the peptide's Asp1 and/or Tyr4 side-chains.
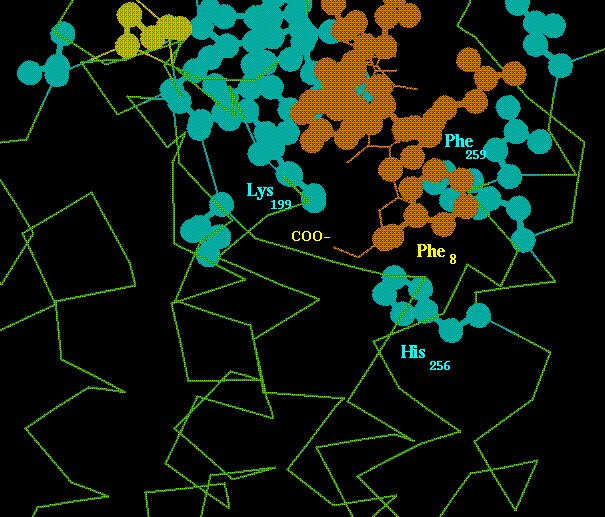
The ligand's C-terminus is found inserted in a transmembrane locus surrounded by helices III, V, VI and VII (Fig. 4), corresponding crudely to the retinal pocket in rhodopsins.
 Click to enlarge
Fig. 4. Interaction of the AngII C-terminus with the AT1 receptor locus which is supposed to trigger receptor activation. The AngII molecule is in brown and receptor residues involved in binding and signaling are in light blue. See the text for more details.
Click to enlarge
Fig. 4. Interaction of the AngII C-terminus with the AT1 receptor locus which is supposed to trigger receptor activation. The AngII molecule is in brown and receptor residues involved in binding and signaling are in light blue. See the text for more details.
The salt-bridge between the peptide's C-carboxylate and the receptor's Lys199(516) side-chain appears to orient a transmembrane mode for docking the aromatic rings of AngII's Phe8 and the receptor's Phe259, which allows the complex formed by the ligand's C-terminal carboxylate and the receptor's Lys199(516) to be placed near the receptor's His256(621) imidazole ring. For this sort of configuration a Phe side-chain at AngII's C-terminal position is required, and the fact that this is also a structural requirement for agonist activity, suggests that the complex of Fig. 4 is crucial for signal triggering mechanisms [15,16].
3. Receptor-G protein coupling
A model describing some general structural features for the formation of a complex between GPCRs and G protein heterotrimer was built [31] based on the following premises:
1. There are about 10 times more types and sub-types of GPCRs than G proteins[1], indicating promiscuity of G proteins in binding to the different receptors [32].
2. The domains of receptors and G proteins involved in reciprocal binding must have conserved features to support the promiscuity. Thus, the conserved residues at the cytosolic side of transmembrane helices, the N-terminal region of loop III-IV and the C-terminal tail are likely to bind G protein. The receptor binding site in transducin's Ga chain is formed by two distinct regions of this protein: a segment of the N-terminal alpha helix; and the last C-terminal amino acids comprising the beta2 strand, the beta2-alpha5 loop, the alpha5-helix and the C-terminal tail with a random structure [33].
3. A great deal of experimental data is available showing that the cytosolic domains and especially loop V-VI of GPCRs are crucial for receptor-G protein coupling. The fact that the structure of this loop is quite variable for receptors involved in the binding to a same G protein trimer indicates that some sort of intermediary structures or adapters must be involved in the coupling. Experimental evidence suggests that cytoskeleton proteins or other modulators might be these special structures [34 ].
4. The structure of the receptor binding site in Galpha chains is invariant but only a few fully conserved residues are found in this region. One of these conserved residues is Asp337 (numbering for Galpha chain residues are in italic), located at the middle of helix alpha5. Interestingly, besides being accessible, this residue is part of a cluster of polar and hydrophobic residues involving discontinuous regions of the Galpha chain [35] which seems to stabilize the alpha5-helix and the GDP binding site of this protein [36,37]. As the major function of GPCRs in coupling to G protein trimer is to trigger a mechanism leading to GDP release, it is conceivable that the receptor might act by binding to the Galpha cluster via Asp337, thus destabilizing the alpha5-helix and the GDP site.
5, Based on GPCRs conservation patterns, we have suggested that Arg126(340), in the DRY motif present at the cystosolic side of helix III, could be the structural element of receptors performing the basic function of interacting with Asp337 in the residue cluster of Galpha chain [17,35].
Fig. 5 shows the complex between AT1 receptor and G protein heterotrimer [31].
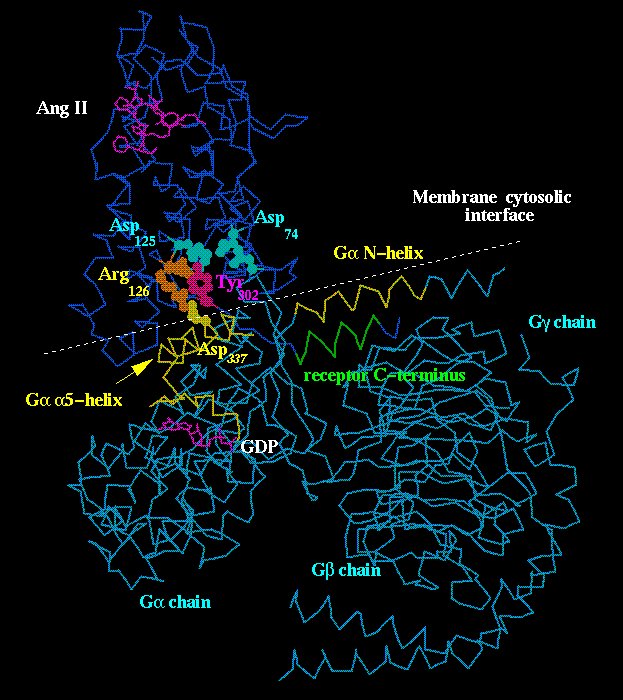 Click to enlarge
Fig. 5. Complex between AT1 receptor and G protein heterotrimer. Note the cytosolic membrane interface which runs near the Galpha N- and Ggamma C-termini (shown in yellow and green, respectively) at which palmitoylation and prenylation sites are present. Arg126(340) at helix III's end is shown in brown for two positions: (1) inactive when pointing towards the receptor's central cavity and interacting with the Asp125(339) side chain (light blue); (2) active when pointing towards Asp337 in the Galpha alpha5-helix (yellow). Tyr302(734) (magenta) is set pointing towards the receptor's Asp125(339) and Asp74(324) (light blue) to simulate the condition which stabilizes the receptor's activated form.
Click to enlarge
Fig. 5. Complex between AT1 receptor and G protein heterotrimer. Note the cytosolic membrane interface which runs near the Galpha N- and Ggamma C-termini (shown in yellow and green, respectively) at which palmitoylation and prenylation sites are present. Arg126(340) at helix III's end is shown in brown for two positions: (1) inactive when pointing towards the receptor's central cavity and interacting with the Asp125(339) side chain (light blue); (2) active when pointing towards Asp337 in the Galpha alpha5-helix (yellow). Tyr302(734) (magenta) is set pointing towards the receptor's Asp125(339) and Asp74(324) (light blue) to simulate the condition which stabilizes the receptor's activated form.
The basic assumptions for the structural matching in this figure are that (1) Arg126(340) at the receptor's helix III could fit Asp337 in the Galpha alpha5-helix; (2) an alpha-helix beginning after the FR motif (positions 750 and 751 in Fig. 1) at the receptor C-terminus could make an antiparallel interaction with the N-helix at the N-terminus of Galpha chain. Arg126(340) is the most conserved residue in the sequence of rhodopsin-like GPCRs [1] and participates in receptor/G protein coupling [18,19]. The GPCR residues around position 750 (Fig. 1) are involved in G protein coupling as described for AT1 receptors [8,10]. That this same region can form an alpha-helix, as depicted in Fig. 5, was suggested by circular dicroism and NMR studies of a peptide possessing the AT1 sequence starting at the position of Tyr302(position 734 in Fig. 1) [38]. As a check for the complex in Fig. 5, it can also be seen that the cytosolic interface of the membrane bilayer, where the receptor's seven-helix bundle is embedded, can be drawn near the palmitoylation and prenylation sites at the extreme ends of the Galpha N-terminus and Ggamma C-terminus.
The model in Fig. 5 allows interpretation of the functioning of GPCRs taking into account two steps:
(1). Inactive state: the Arg126(340) side-chain would be pointing towards the receptor central cavity and perhaps interacting with the Asp125(339) side-chain;
(2) Activated state: agonist binding or light absorption (for opsins), could initiate the receptor signaling which propagates to the cytosolic side of the receptor, resulting in protonation of Asp125(339) and Asp74(224) [39,40]; this would cause the Arg126(340) side-chain to move towards Asp337 of the Galpha chain, triggering the mechanism of GDP release and G protein activation.
One of the most interesting events occurring in GPCR-G protein coupling is the increase in the receptor's affinity for agonist or the stabilization of the receptor-activated state [41]. A major role of transducin Galpha chain C-terminus in this mechanim has been supported experimentally since a peptide with the sequence of this region can by itself stabilize metarhodopsin II becoming an alpha-helix in the course of this event [42]. The C-terminal end of the Galpha chain is not shown in the model of Fig. 5 but one can speculate, taking into account the sizes of the structures involved, that this G protein segment might interact with the receptor's C-terminus thus acquiring the alpha-helix configuration. Due to this mechanism, Tyr302(734), at the begining of the receptor's C-terminal tail, could point towards the receptor central cavity to interact with Asp74(224) and Asp125(339), instead of Arg126(340), and thus to stabilize the activated receptor state (Fig. 5). This role for Tyr302(734) is also supported experimentally since mutation of this residue is found to impair receptor/G protein coupling and receptor phosphorylation and internalization, an event that occurs only with activated receptor [22,43]
| <= Materials & Methods | RESULTS | Discussion & Conclussions => |
| Discussion Board | Next Page | Your Symposium |