Cell Biology Poster Session
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Gene
Therapy for a Murine Model of a Genetic Lysosomal Storage Disease with
Cell Microencapsulation.
Contact Person: Colin JD Ross (rosscj@mcmail.cis.mcmaster.ca)
Introduction
Overview – This research investigates how to deliver a gene to the brain to treat a CNS disorder.
Mucopolysaccharidosis type VII (MPS VII) is caused by a deficiency of the lysosomal enzyme beta-glucuronidase. Clinical manifestations include intralysosomal accumulation of undegraded glycosoaminoglycans. We propose an alternative treatment strategy, by implanting allogeneic cells enclosed in biocompatible & perm-selective microcapsules that allow the diffusion of nutrients, waste and therapeutic products, while excluding immune mediators. Since the cells are immuno-protected, the capsules may then be implanted into different patients requiring the same gene product, making this a cost-effective therapeutic approach.
I. Fibroblasts expressing mouse beta-glucuronidase enclosed in microcapsules were implanted intraperitoneally into MPS VII mice (3-5 ml). Enzyme was detected in the plasma (up to 66% of physiological level), in the kidney for up to 4 weeks, and in the liver and spleen for up to 8 weeks. Tissue sections showed a reduction in lysosomal storage lesions in the spleen and pathology reversal of the liver for up to 8 weeks. However, implanted mutant mice developed antibodies against the recombinant beta-glucuronidase, preventing long-term delivery in vivo, evidencing the difficulties to deliver therapeutic products to individuals with a null mutation and when the enzyme cannot cross the blood brain barrier to the CNS.
II. Recently, we have explored directly implanting microcapsules into the CNS (5 microlitres) for treatment of the neurodegeneration. We have been able to deliver high levels of beta-glucuronidase, and reduce or clear disease pathology throughout the CNS for up to 8 weeks.
III. Furthermore, in a pilot study we have combined CNS (1 microlitre) + peripheral (1 ml) treatments, for therapy of this diseases throughout the entire body of the MPS VII mouse.
Some more background - The genetic lysosomal storage disease mucopolysaccharidosis type VII is a deficiency of beta-glucuronidase. This enzyme breaks down glycosaminoglycans, and a deficiency can lead to glycosaminoglycan accumulations in cells causing progressive neurodegenration and early death (4-10 years). Although MPS VII is rare, it is an excellent disease for study because: (1) MPS VII is a very simple and well understood disease, and an excellent starting point upon which to develop novel treatments. (2) MPS VII is an excellent model of neurodegeneration. (3) There is an good animal model of MPS VII that closely resembles the human disease, which has been used to asses a variety of gene delivery methods (references 2-5). (4) More common diseases, such as Tay-Sach’s and Gaucher’s disease, are almost identical to MPS VII. (5) There is a broad spectrum of neurological diseases that would benefit from an increased understanding of gene delivery to the brain such as single gene disorders (Huntington’s disease) and multifactorial diseases (ie. Alzheimer’s, schizophrenia, and cancer).
To develop a cost-effective approach of enzyme delivery, we initiated in our laboratory a novel form of gene therapy. A universal cell line is genetically modified to produce high levels of a desired enzyme. Cells are implanted into patients requiring the same enzyme. To avoid host rejection, cells are enclosed in small (<1 mm) immuno-isolation devices that protect from rejection, but allow cell survival and produce a supply of the therapeutic enzyme for the patient (See overview figure 1)
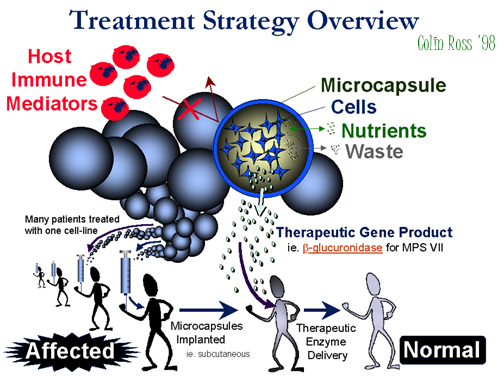 Fig.
A:Overview.
Fig.
A:Overview.
Previously we examined the delivery of beta-glucuronidase to MPS VII mice, which was successful for delivery to peripheral organs (data not shown), but the enzyme could not reach the brain because of the blood-brain barrier. To overcome this barrier, we previously examined directly injecting microcapsules into the brain which were producing a “marker” gene, which was easily detected in the brain for at least sixteen weeks post-implantation(Ross et al, 1999).
Back
to the top.
Materials
and Methods
CELLS ENCAPSULATION.
Cells harvested, suspended in 1.5% alginate, and extruded into 1.1% CaCl2 (Fig 9) followed by washes in 0.28% CaCl2, 0.1% CHES, 1.1% CaCl2, 0.05% PLL, 0.1% CHES, 1.1% CaCl2, 0.9% NaCl, 0.03% alginate, 0.9% NaCl, 0.055M sodium citrate, and media. Resulting CNS capsules ~150 micrometer diameter, peripheral capsules ~500 micrometer.
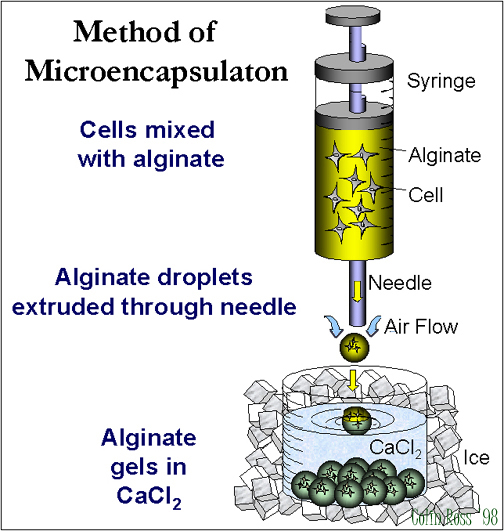 Fig.
B: Encapsulation.
Fig.
B: Encapsulation.
MPS VII MICE
bred at the University of Toronto (Dr. Ralph) were treated at U of T or shipped to McMaster University for treatment. Mice were genotyped with a PCR based-assay that we developed.
MICROCAPSULE IMPLANTATION
Mice anesthetized and implanted peripherally: intraperitoneal (3-5 ml), or subcutaneous (1 ml) with a 16 gauge needle and/or intraventricullarily into CNS (1-5 µl) with a glass micropipette through a burr hole drilled into skull.
IN VIVO GENE PRODUCT DELIVERY
Organs or CNS removed, sectioned, homogenized, spun, and supernatant assayed for beta-glucuronidase (4-MU). Total protein content measured with Lowry or Bio-Rad Assays.
HISTOLOGY
Histlogy examined with toluidine blue stained 500 nm sections fixed and embedded in Spurr resin, photographed under light microscopy with a Zeies Axioskop 160-1000x magnification.
ANTIBODY ASSAY
Anti-beta-glucuronidase antibodies assayed with a method we developed: unpurified enzyme of known amount mixed with blood samples, then samples immunoprecipitated, and enzyme-antibody complexes spun down and removed, remaining enzyme quantified with 4-MU ?-glucuronidase assay as an inverse measure of antibody titre
Back
to the top.
Results
OVERVIEW-I
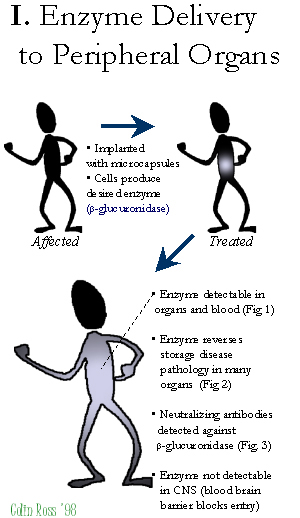 Fig.
C: Overview-I.
Fig.
C: Overview-I.
PERIPHERAL treated MPS VII mice implanted with microcapsules show high enzyme delivery detectable in organs and blood (Fig. 1) resulting in reduced of lysosomal storage disease pathology in peripheral organs (Fig. 3), although neutralizing antibodies are detected (Fig. 2) and the blood brain barrier inhibits enzyme delivery to the CNS.
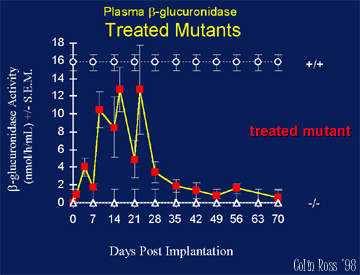 Fig.
1: Plasma.
Fig.
1: Plasma.
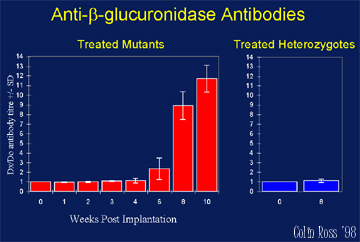 Fig.
2: Antibodies.
Fig.
2: Antibodies.
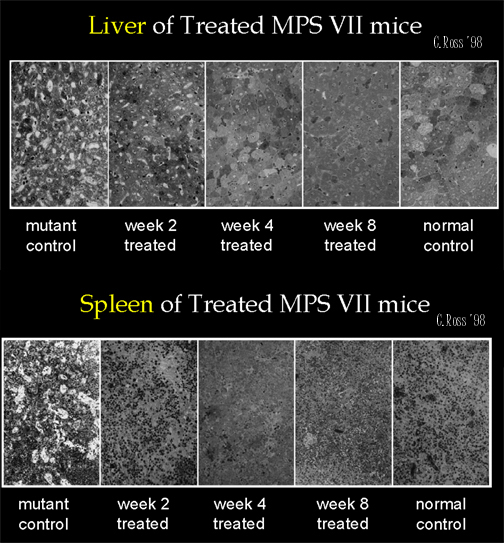 Fig.
3: Organs Histology.
Fig.
3: Organs Histology.
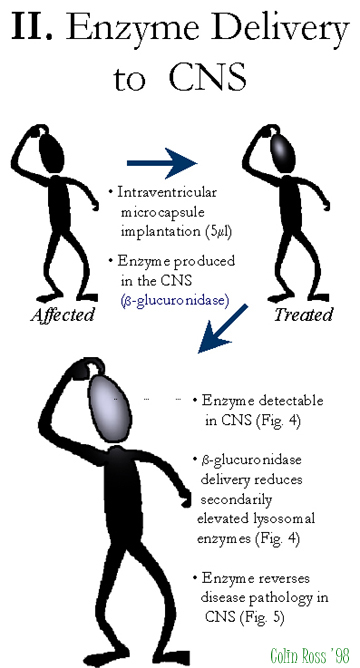 Fig.
D: Overview-II.
Fig.
D: Overview-II.
CNS treated MPS VII mice, intraventricularily treated with 5 ?l of microcapsules producing beta-glucuronidase show high levels of delivery to the CNS (Fig. 4) with reduced lysosomal storage disease pathology in the CNS (Fig 5). We have seen delivery of beta-glucuronidase directly into the brain of MPS VII mice for up to 8 weeks (longest time-point examined) with the highest levels are near the implantation site with positive results biochemically & histologically.
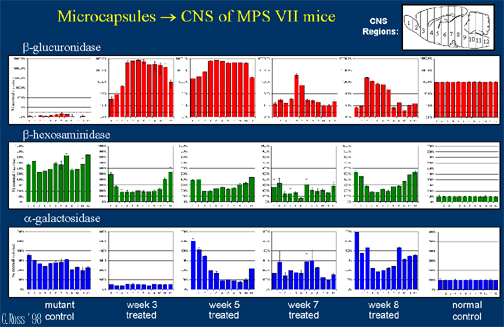 Fig.
4:CNS Enzyme.
Fig.
4:CNS Enzyme.
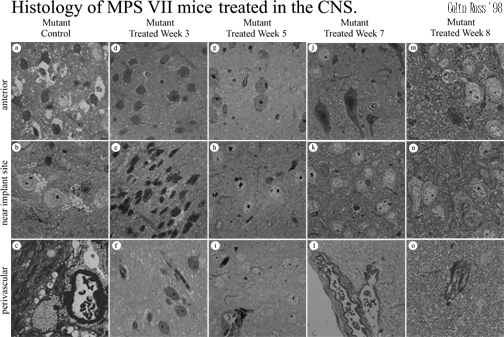 Fig.
5:CNS histology.
Fig.
5:CNS histology.
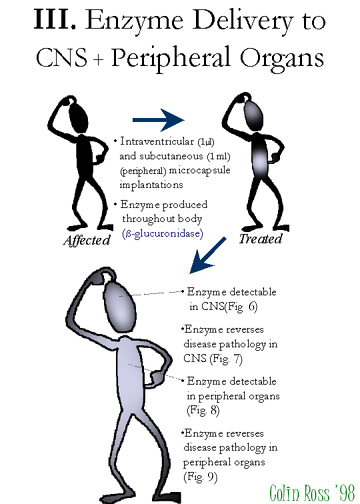 Fig.
E:Overview-III.
Fig.
E:Overview-III.
A pilot study with 5-fold lower doses in both CNS+peripheral sites has shown similar results, MPS VII mice treated in both the CNS (1 microlitre intraventricularily) and peripherally (subcutaneously 1 ml) show high levels of beta-glucuronidase delivery to the CNS (Fig. 6) and peripheral organs such as kidney, liver, spleen, and muscle (Fig 8), with reduced lysosomal storage disease pathology in the CNS (Fig 7) and peripheral organs (Fig. 9).
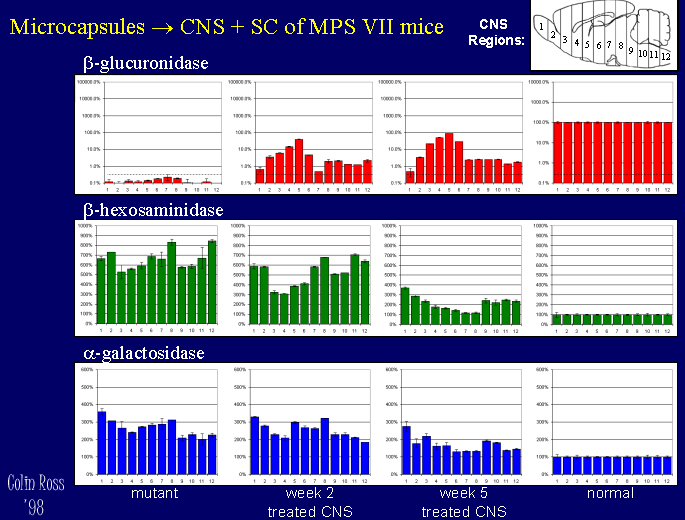 Click to enlarge
Click to enlarge
Fig. 6: CNS+Peripheral - CNS Enzyme.
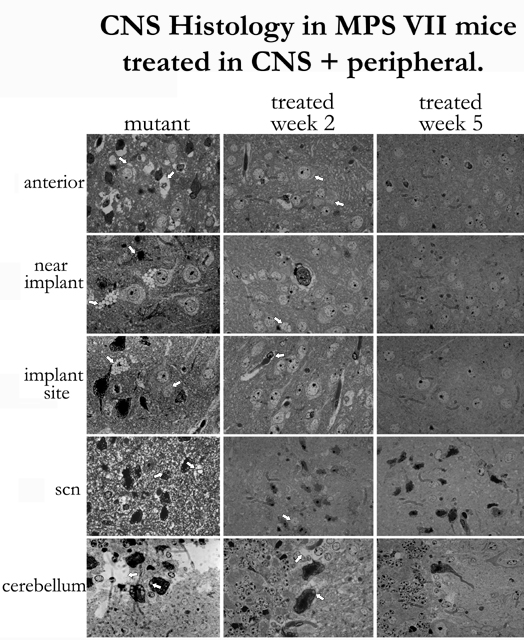 Fig.
7: CNS+Peripheral - CNS Histol
Fig.
7: CNS+Peripheral - CNS Histol
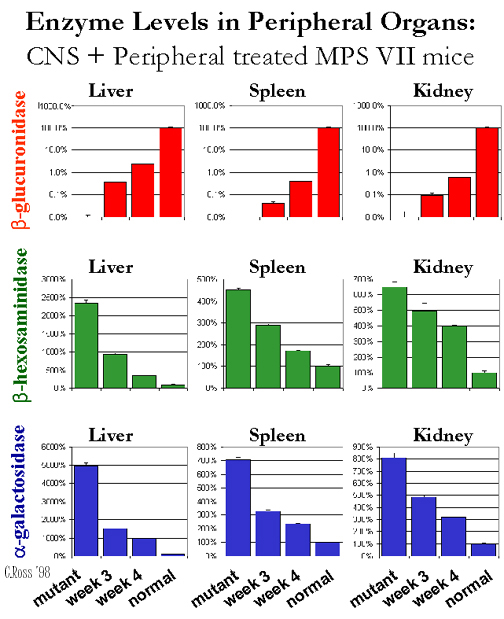 Fig.
8: CNS+Peripheral - Organs Enz.
Fig.
8: CNS+Peripheral - Organs Enz.
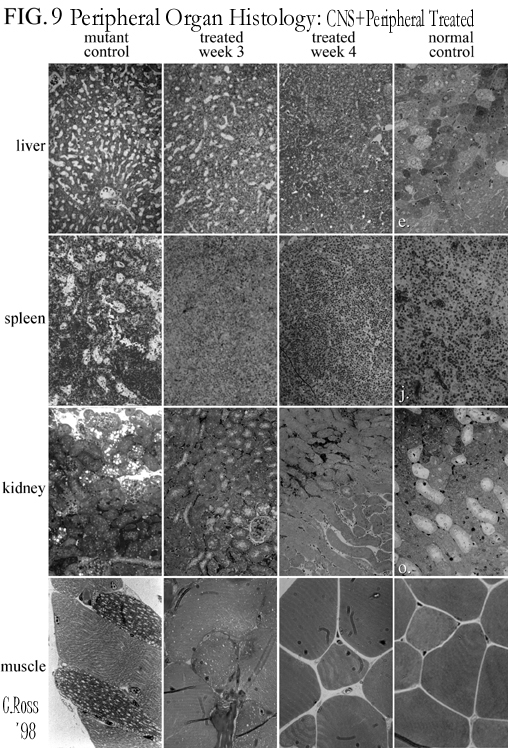 Fig.
9:CNS+Peripheral - Organs Histol.
Fig.
9:CNS+Peripheral - Organs Histol.
Back
to the top.
Discussion
and Conclusion
1. Encapsulated cells producing high levels of beta-glucuronidase (2A50 fibroblasts courtesy Dr. W.Sly) were implanted into the abdominal cavities of MPS VII mice. beta-glucuronidase was detected in plamsa (up to 66% of normal) and significant levels also detected in kidney, liver, and spleen.
2. Anti-beta-glucuronidase antibodies detected, and beta-glucuronidase was not detected in the CNS due to the blood brain barrier.
3. By implanting directly into the brain, beta-glucuronidase was delivered at high levels throughout the CNS, for > 8 weeks.
4. Secondarily elevated enzymes reduced (beta-hexosaminidase and alpha-galactosidase)
5. Lysosomal disease storage pathology is reduced
6. We combined the two treatment sites for therapeautic benefit to both the CNS & peripheral organs (pilot study).
7. These results show the therapeautic effects possible from implantable microcapsules as a gene delivery system
Back
to the top.
References
- Ross C. J.D., Ralph M. & Chang, P.L. Delivery of recombinant gene products to the CNS with non-autologous cells in alginate microcapsules. Human Gene Therapy, 10 (1):(in press), 1999.
- Wolfe, J. H., S. L. Deshmane & N. W. Fraser. Herpesvirus vector gene transfer and expression of beta-glucuronidase in the central nervous system of MPS VII mice. Nat Genet. 1, 379-84. 1992.
- Snyder, E.Y., Taylor, R.M. & Wolfe, J.H. Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature. 374, 367-70. 1995.
- Ohashi, T., Watabe, K., Uehara, K., Sly, W.S., Vogler, C., & Eto, Y. Adenovirus-mediated gene transfer and expression of human beta-glucuronidase gene in the liver, spleen, and central nervous system in MPS VII mice. Proc Natl Acad Sci U S A. 94:1287-1292. 1997.
- Taylor, R.M., & Wolfe, J.H. Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector-corrected fibroblasts secreting high levels of beta-glucuronidase. Nat Med. 3, 771-774. 1997.
Back
to the top.
| Discussion
Board | Previous
Page | Your
Poster Session |