Invited Symposium: Na-H Exchangers and Intracellular pH Regulation
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
VASCULAR DISEASE IN DIABETES
Biochemical abnormalities of glucose metabolism leading to hyperglycemia are occurring in a significant and increasing number of people. Type I diabetes is most usually an auto-immune disorder which occurs early in life and requires insulin for the individuals survival. Type II diabetes occurs later in life and is associated with an insulin-resistant state.
The hyperglycemia of the latter state may be treated by drugs, which release or increase the sensitivity of tissues to insulin, or by administration of exogenous insulin. However, in both situations the major pathological manifestations that accompany the primary diabetes, referred to as "complications", are most pronounced in the vasculature.
Vascular disease is conveniently distinguished into categories of micro- and macro-vascular disease. Micro-vascular disease is associated with retinopathy, nephropathy and neuropathy and thus diabetes becomes a significant contributor to renal failure and blindness [1]. Micro-vascular disease occurs as a result of chronic hyperglycemia and involves vascular derived growth factors [2, 3]. Macro-vascular disease manifests as accelerated atherosclerosis of coronary and peripheral arteries and is a major cause of heart attacks and amputations [4].
The obvious candidates that may mediate vascular disease in diabetes are the hyperglycemia, hyperinsulinemia and dyslipidemia, with possible compounding interactions due to hypertension. The recent United Kingdom Prospective Diabetes Trial [5] confirmed the role of hyperglycemia in microvascular disease and has demonstrated a trend towards beneficial macrovascular effects from lowering blood glucose either through administration of oral hypoglycemic agents or insulin. These data suggest that the hypoglycemic action of insulin outweights any direct pro-atherosclerotic actions. In contrast to treatments for hypertension and hypercholesterolemia in which the abnormalities can be normalized, the UKPDS data showed an increasing hyperglycemia over time with diabetes in both the normal and intensively treated groups [5]. Hence the full impact of hyperglycemia on macrovascular complications may not become apparent until therapeutic modalities are available which produce long term normoglycemia.
One of the primary regulators which has been extensively implicated in various vascular disease states is the plasma membrane sodium-hydrogen exchanger. Many studies have implicated this transporter in cell proliferation since it serves as a point of coalescence for growth factor signals. Many biochemical abnormalities of the diabetic state have the potential to regulate or modulate NHE activity and these are considered below.
SODIUM/HYDROGEN EXCHANGER: STRUCTURE, FUNCTION AND REGULATION
The sodium hydrogen exchanger (NHE) is a 110 kDa integral plasma membrane glycoprotein with 10 or 12 putative membrane spanning regions. Five isoforms of NHE (NHE-1 to 5) have been identified which all have a similar structure [6, 7] but vary in molecular weight, level and type of glycosylation, sensitivity to inhibitors, and cell or tissue distribution [8, 9]. NHE-1 is ubiquitously expressed, whilst other isoforms have more limited ranges of expression and thus may perform specialized functions [6].
NHE functions as an exchanger of intracellular acid for extracellular sodium and as such maintains ionic homeostasis, in particular intracellular pH. However, interest in NHE increased dramatically when its activity was also shown to be regulated by hormones and growth factors including platelet derived growth factor (PDGF), angiotensin II (AII), neurotransmitters, chemotactic peptides and thrombin [9-11]. These studies provided the first evidence that rather than just a homeostatic role, NHE-1 might also actively contribute to the regulation of cell growth and/or proliferation. In agreement with this a number of studies have demonstrated that pharmacological NHE-1 inhibitors block VSMC (vascular smooth muscle cell) proliferation in vitro [12] and in vivo [13, 14]. Moreover, overexpression of NHE-1 was shown to increase proliferation of VSMC [15].
In contrast to initial speculation and some data, the mechanisms by which these growth factors regulate NHE-1 activity have proven to be complex. Potential regulatory levels include post-translational modifications, (e.g., phosphorylation), changes in expression levels (altered rates of transcription or translation) and alterations in the association of NHE-1 with regulatory proteins such as calmodulin (CaM) and calcineurin homologous protein (CHP), or with ATP. These individual regulatory mechanisms have been reviewed recently elsewhere [16].
BIOCHEMICAL ABNORMALITIES OF DIABETES AND NHE IN VASCULAR SMOOTH MUSCLE
The potential for the diabetic milieu to modulate the functions of vascular smooth muscle and endothelial cells has been recognized for a long time. Such functions include the proliferation, apoptosis and migration of VSMC or endothelial cells and additionally tube formation in microvascular endothelial cells. Matrix production and degradation is also an important function of both cell types [17] but little is known of the role of NHE in the regulation of this function.
The literature consistently reports that hyperglycemia stimulates proliferation of VSMC however, its effects on endothelial cell proliferation are unclear. This latter confusion may derive from the multitude of diverse endothelial cell models which have been employed, particularly differences between micro- and macrovascular endothelial cells and possibly because endothelial cells have undergone extensive proliferation before the experimental period. Additionally, the multitude of experimental designs employing either acute or chronic treatment with hyperglycemia coupled with the use of multiple concentrations of growth factor stimulation adds a further layer of complexity. The effects of key diabetic abnormalities on vascular cell function and the proven or potential involvement of NHE, is considered below.
a) Insulin/Insulin-like growth factor-1: Insulin and insulin-like growth factor I (IGF-1) receptors have been identified on VSM and endothelial [18] cells and can cross react, although the difference in their affinities is over an order of magnitude. These agonists elicit signaling cascades that are dependent on their relative concentrations and have not been fully described. IGF-1 is interestingly also produced by vascular cells and may therefore be more relevant as an autocrine factor compared to the paracrine action of insulin [18, 19].
Insulin and IGF-1 stimulate migration and tube formation but not the proliferation of bovine carotid endothelial cells [20]. However, these agonists have been shown to stimulate proliferation of VSMC [21, 22] and this may be mediated by NHE-1. Insulin has also been shown to regulate NHE-1 activity and expression in non-vascular wall cells [23-26] and IGF-1 regulates NHE-1 activity in vascular cells such as those in mesenteric arteries [27] and rat aorta [28]. However, a firm role and the signaling pathway(s) involved in insulin and IGF-1 regulation of NHE-1 and thus vascular cell growth function is still to be established.
b) Hyperglycemia: Hyperglycemia is now recognized as a significant causative factor in microvascular disease and its association with macrovascular disease is strong but not yet proven. Hyperglycemia elicits a variety of well defined cellular changes and these primary effects have a number of secondary consequences which have not been fully characterized. The primary biochemical effects of hyperglycemia relevant to regulation of NHE-1 include: i) de novo synthesis of diacylglycerols (DAG); ii) increased oxidative stress; and iii) elevated production of advanced glycation end products. The secondary biochemical effects mediated by hyperglycemia include increased growth factor production and release.
(i) de novo synthesis of DAG: Hyperglycemia increases the synthesis of diacylglycerol (DAG) by enhancing the glycolytic production of DAG precursors. DAGs are formed by interactions between glucose derivatives and fatty acids. The major role of DAGs is activation of specific protein kinase C (PKC) isoforms (e.g. alpha and beta) which causes a phosphorylation signaling cascade that has effects on cell metabolism and gene expression. [29]. PKC activates NHE activity in almost all cells studied including vascular cells [30]. The actions of DAG are terminated by DAG kinase, an enzyme that is activated by the anti-oxidant, vitamin E.
(ii) Increased oxidative stress: Hyperglycemia directly increases oxidative stress (reactive oxygen species or free radicals) in a number of cell types including VSMC and endothelial cells [31-33]. Typically this is achieved via the auto-oxidation of sugars and nonenzymatic glycation products. For example, hyperglycemia or the growth factors AII, PDGF, TGFb stimulate production of an F2-isoprostane, 8-epiprostaglandin F2 alpha, from free-radical-catalyzed peroxidation of arachidonic acid, which subsequently stimulated VSMC proliferation [34]. Hyperglycemia induced apoptosis of human umbilical endothelial cells by a process not involving PKC. It was suggested that the production of oxygen free radicals by autooxidative processes instead lead to the activation of transcription factors necessary for apoptosis [35].
Diabetes is associated with hyperlipidemia and elevated levels of atherogenic lipoproteins, including oxidized LDL. Oxidized LDL activates vascular proliferation by stimulating MAPK in VSMC but not in endothelial cells [36]. This suggests that oxidized LDL may activate NHE in VSMC. However, native and minimally oxidized LDL actually inhibit NHE activity in platelets [37]. The role of LDL and its derivatives on regulating NHE activity in VSMC requires further investigation.
(iii) Advanced glycation end products (AGEs): Glucose reacts with proteins, such as collagen and BSA, to form reversible early glycosylation products (Schiff bases) at a rate proportional to the glucose concentration; these intermediates rearrange to form more stable but also reversible Amodori products. The Amodori products rearrange to form irreversible end products known AGEs. AGEs bind to their receptor, RAGE (receptor for AGE), and exert its effect directly on the cell. AGEs initiate three primary effects on vascular tissue [38]: firstly, activation of gene expression, such as growth factors (IGF-1, TGFb1, PDGF) or extracellular matrix proteins (fibronectin, laminin, collagen IV) [39, 40]; secondly, alteration of signaling through modifications of extracellular matrix molecules [41]; and thirdly, direct modifications due to intracellular AGEs reacting with target proteins. Alternatively AGEs react with the extracellular matrix, bind and accumulate on the vessel walls resulting in thickened, inelastic vessel walls increasing the risk of vascular complications [38].
Interestingly, a recent publication reported that AGEs stimulate MAP kinase activity and proliferation of rabbit VSMC but not endothelial cells [42]. This proliferation may have been due to the secondary effects of AGEs increasing secretion of growth factors such as PDGF or by inducing generation of reactive oxygen intermediates [38].
Adhesion of VSMCs induces activation of PKC and primes the cells for a mitogenic response. Adhesion also stimulates NHE activity and thus elevates intracellular pH [43]. This process is mediated by interactions between cell surface integrins and extracellular matrix molecules, in particular fibronectin. However, glycation of fibronectin inhibits attachment and proliferation of human VSMCs [41]. The effect of glycated fibronectin on NHE activity is unknown but one can speculate that it will be inhibitory; this is interesting in that protein modifications are often presumed to be deleterious but here glycation appears to contribute to vascular quiescence. Nevertheless, impaired adhesion may, for example, promote migration, an early response in the process of restenosis following coronary angioplasty.
(iv) Mechanistically uncharacterized effects of hyperglycemia: In contrast to the ubiquitous studies demonstrating stimulation of VSMC proliferation mediated by hyperglycemia, the reported effects on endothelial cells are mixed and very dependent upon experimental conditions [36, 44-47]. Hayashi et al [48] showed hyperglycemia enhanced proliferation of bovine carotid artery endothelial cells under low serum stimulation but inhibited in the presence of 10% serum. We have recently examined this question in bovine aortic endothelial cells. Using endogenous proliferation in the absence of serum or a submaximal concentration of serum (2%) to allow for stimulation or inhibition, we found that under both conditions hyperglycemia inhibited the rate of proliferation (Figure 1) which confirms a number of other findings [44, 45, 47]. We have also demonstrated that the NHE inhibitor HOE 694 inhibits proliferation of endothelial cells grown under standard cell culture conditions implying a role for NHE as we have previously reported in VSMC [12].
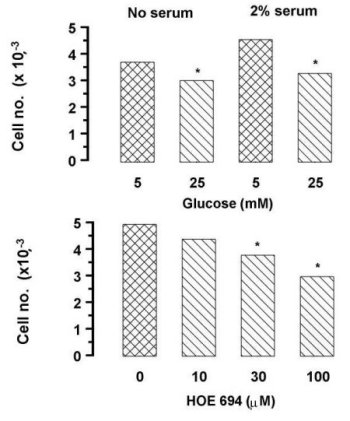 Fig. 1: Effects of hyperglycemia
and sodium-hydrogen exchange inhibition on macrovascular endothelial cell
proliferation. The upper panel shows an increase in bovine aortic endothelial
cell numbers over two days exposed to normo- or hyperglycemic conditions
in the presence of absence of a half maximal (2%) stimulatory concentration
of serum. The lower panel shows the effect of HOE 694 on cells grown over
two days under normoglycemic conditions plus 2% serum. The cells were washed,
attached cells dislodged with trypsin/versene and counted in a Coulter
Counter. Data are from two experiments in triplicate (* p<0.05).
Fig. 1: Effects of hyperglycemia
and sodium-hydrogen exchange inhibition on macrovascular endothelial cell
proliferation. The upper panel shows an increase in bovine aortic endothelial
cell numbers over two days exposed to normo- or hyperglycemic conditions
in the presence of absence of a half maximal (2%) stimulatory concentration
of serum. The lower panel shows the effect of HOE 694 on cells grown over
two days under normoglycemic conditions plus 2% serum. The cells were washed,
attached cells dislodged with trypsin/versene and counted in a Coulter
Counter. Data are from two experiments in triplicate (* p<0.05).
We also examined the acute effect of hyperglycemia on chemokinetic (migratory)
activity of endothelial cells in a scrape/wound assay. Under these conditions
hyperglycemia did not influence the extent of endothelial cell migration
(Figure 2).
 Fig. 2: Effects of hyperglycemia
on the chemokinetic activity of macrovascular endothelial cells. Bovine
aortic endothelial cells were grown to confluency then serum deprived for
24 h, subjected to a scrape injury (Top) and cultured for a further 20
h under norm- (Middle) or hyperglycemic (Lower) conditions. Cells were
paraformaldehyde fixed, stained with Mayer's Hematoxylin and computer imaged
for determination of migratory activity.
Fig. 2: Effects of hyperglycemia
on the chemokinetic activity of macrovascular endothelial cells. Bovine
aortic endothelial cells were grown to confluency then serum deprived for
24 h, subjected to a scrape injury (Top) and cultured for a further 20
h under norm- (Middle) or hyperglycemic (Lower) conditions. Cells were
paraformaldehyde fixed, stained with Mayer's Hematoxylin and computer imaged
for determination of migratory activity.
This result is in contrast to the finding of Gade et al [49] who demonstrated an inhibition of migration in an in vitro model. There are few studies on the effects of hyperglycemia on NHE in endothelial cells. Canessa and colleagues demonstrated an inhibitory effect of hyperglycemia on NHE activity occurs without an effect on NHE-1 mRNA levels indicating that the effect is due to translational or post-translational modification [50]. Interestingly, similar effects were observed in response to prolonged hypoxia but in this latter case NHE protein was elevated [51]. (v) Secondary effects of hyperglycemia - growth factor production: Hyperglycemia may have secondary effects by altering growth factor production and secretion. Hyperglycemia, possibly via PKC, stimulates the release of TGFb (and possibly other growth factors) from VSMC [52, 53] but not endothelial cells [54]. TGFb in turn activates signaling pathways in VSMC and stimulates growth and extracellular matrix production [53].
In a number of cases hyperglycemia, via primary or secondary mechanisms, stimulates VSMC growth [34, 38, 44, 53] which may be mediated by NHE-1. In fact, hyperglycemia stimulated NHE-1 mRNA in VSMC [55]. These experiments were conducted in the presence of 10% serum indicating that hyperglycemia potentiates the action of growth factors rather than being mitogenic per se. Another study demonstrated an increase in NHE-1 activity, but a confounding decrease in NHE-1 phosphorylation in vascular cells [56]. In a number of cases a role for DAG-induced PKC signaling has been implicated in the regulation of NHE-1 activity in VSMC [11, 55]. On the other hand the effect of oxidative stress, induced by hyperglycemia, on NHE-1 activity has only been examined in neonatal cardiac myocytes. In this study free radicals stimulated MAPK and NHE-1 activity [57]. In the case of hyperglycemia induced AGEs there have been no studies examining a role for NHE-1. Secondary effects of hyperglycemia may also be mediated by the released growth factors regulating NHE-1 activity and thus promoting VSMC growth.
These studies suggest a possible role for NHE-1 regulation of VSMC and endothelial cell proliferation during hyperglycemia and other conditions associated with diabetes.
CONCLUSIONS
Diabetes represents a state of accelerated pathogenesis in the cardiovascular system. A considerable body of evidence exists linking activation of NHE with vascular activation suggesting that diabetes may be a useful condition in which to examine the role of this transporter in vascular pathology. Vascular smooth muscle and endothelial cells possess NHE which are structurally identical but probably differ in their regulatory mechanisms. NHE has been implicated in the functioning of these cells and thus modulation by either direct antagonists or drugs interfering with regulatory pathways is possible. As both vascular smooth muscle and endothelial cells contribute to adaptive and pathophysiological vascular remodeling, in vivo studies are required to unravel the therapeutic potential of NHE antagonists in preventing vascular disease.
Acknowledgments: Work described in this report was supported by a grant-in-aid from the National Heart Foundation of Australia (PJL) and a Block Institute Grant to the Baker Medical Research Institute from the National Health and Medical Research Council of Australia.
References
1. Derkert, T., J.E. Poulsen, and M. Larsen, Prognosis of diabetes onset before the age of thirty-one. II. Factors influencing the prognosis. Diabetaologia, 1978. 14(6): p. 371-377.
2. Diabetes Control Complications Trial Research Group, The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The New England Journal of Medicine, 1993. 329(14): p. 977-986.
3. Pfeiffer, A. and H. Schatz, Diabetic microvascular complications and growth factors. Experimental and Clinical Endocrinology and Diabetes, 1995. 103: p. 7-14.
4. Jensen-Urstad, K.J., et al., Early atherosclerosis is retarded by improved long-term blood glucose control in patients with IDDM. Diabetes, 1996. 45(9): p. 1253-1258.
5. UK Prospective Diabetes Study (UKPDS) Group, Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). The Lancet, 1998. 352: p. 837-853.
6. Counillon, L. and J. Pouyssegur, Structure-function studies and molecular regulation of the growth factor activatable sodium-hydrogen exchanger (NHE-1). Cardiovasular Reasearch, 1995. 29: p. 147-154.
7. Ikeda, T., et al., Identification of cytoplasmic subdomains that control pH-sensing of the Na+/H+ exchanger (NHE-1): pH-maintenance, ATP-sensitive, and flexible loop domains. Journal of Biochemistry, 1996. 121: p. 295-303.
8. Yun, C.H.c., et al., Mammalian Na+/H+ exchanger gene family: structure and function studies. American Journal of Physiology, 1995. 269(32): p. G1-G11.
9. Siffert, W. and R. Dusing, Na+/H+ exchange in hypertension and in diabetes mellitus - facts and hypotheses. Basic Research in Cardiology, 1996. 91: p. 179-190.
10. Rao, G.N., et al., Differential regulation of Na+/H+ antiporter gene expression in vascular smooth muscle cells by hypertrophic and hyperplasic stimuli. The Journal of Biological Chemistry, 1990. 265(32): p. 19393-19396.
11. Mitsuka, M. and B.C. Berk, Long-term regulation of Na+/H+ exchange in vascular smooth muscle cells: role of protein kinase C. American Journal of Physiology (Cell Physiology 29), 1991. 260: p. C562-C569.
12. Bobik, A., et al., Ethyisopropylamiloride-sensitive pH control mechanisms modulate vascular smooth muscle cell growth. American Journal of Physiology, 1991. 260(29): p. C581-C588.
13. Kranzhofer, R., et al., Suppression of neointimal thickening and smooth muscle cell proliferation after arterial injury in the rat by inhibitors of Na(+)-H+ exchange. Circulation Research, 1993. 73(2): p. 264-268.
14. Mitsuka, M., M. Nagae, and B.C. Berk, Na(+)-H+ exchange inhibitors decrease neointimal formation after rat carotid injury. Effects on smooth muscle cell migration and proliferation. Circulation research, 1993. 73(2): p. 269-275.
15. Takewaki, S.-i., et al., Activation of Na+/H+ antiporter (NHE-1) gene expression during growth, hypertrophy and proliferation of the rabbit cardiovascular system. Journal of Molecular and Cellular Cardiology, 1994. 27: p. 729-7422.
16. Hannan, K.M. and P.J. Little, Mechanisms regulating vascular smooth muscle Na/H exchanger (NHE-1) in diabetes. Biochemistry and Cell Biology, 1998. submitted.
17. Spiro, M.J. and M.L.D. D'Autilia, Effect of high glucose on formation of extracellular matrix components by cultured rat heart endothelial cells. Diabetologia, 1995. 38: p. 430-436.
18. Bar, R.S., et al., Insulin, insulin-like growth factors, and vascular endothelium. American Journal of Medicine, 1988. 85(5A): p. 59-70.
19. Sowers, J.R., Effects of insulin and IGF-1 on vascular smooth muscle glucose and cation metabolism. Diabetes, 1996. 45: p. S47-51.
20. Nakao-Hayashi, J., et al., Stimulatory effects of insulin and insulin-like growth factor I on migration and tube formation by vascular endothelial cells. Atherosclerosis, 1992. 92(2-3): p. 141-149.
21. Begum, N., et al., Vascular smooth muscle cell growth and insulin regulation of mitogen-activated protein kinase in hypertension. American Journal of Physiology, 1998. 275(1): p. C42-C49.
22. Cruzado, M., et al., Proliferative effect of insulin on cultured smooth muscle cells from rat mesenteric resistance vessels. American Journal of Hypertension, 1998. 11(1): p. 54-58.
23. Semplicini, A., et al., Posttranslational effects of prtoein kinase C and insulin on red cell membrane phosphorylation and cation heteroexchange in hypertension. Blood Pressure, 1996. 1: p. 55-58.
24. Ceolotto, G., et al., Protein kinase C and insulin regulation of red blood cell Na+/H+ exchange. American Journal of Physiology, 1997. 272(3): p. C818-C826.
25. Incerpi, S., et al., Insulin stimulation of Na/H antiport in L-6 cells: a different mechanism in myoblasts and myotubes. Journal of Cell Physiology, 1997. 171(3): p. 235-242.
26. Johnson, D., et al., Insulin-like growth factor I stimulates apical sodium/hydrogen exchange in human proximal tubule cells. American Journal of Physiology, 1997. 272(4): p. F484-F490.
27. Touyz, R.M. and E.L. Schiffrin, Growth factors mediate intracellular signalling in vascular smooth muscle cells through protein kinase C-linked pathways. Hypertension, 1997. 30(6): p. 1440-1447.
28. Standley, P.R., et al., IGF-1 regulation of Na(+)-K(+)-ATPase in rat arterial smooth muscle. American Journal of Physiology, 1997. 273(1): p. E113-E121.
29. Ishii, H., et al., Amelioration of vascular dysfunctions in diabetic rats by an oral PKC b inhibitor. Science, 1996. 272: p. 729-721.
30. Kitazone, T., et al., Involvement of calcium and protein kinase C in the activation of the Na+/H+ exchanger in cultured bovine aortic endothelial cells stimulated by extracellular ATP. Biochemical and Biophysical Acta, 1989. 1013(2): p. 152-158.
31. Guijarro, C., et al., 3-Hydroxy-3-methylglutaryl coenzyme A reducdase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circulation Research, 1998. 83: p. 490-500.
32. Giugliano, D., A. Ceriello, and Paolisso, Oxidative stress and diabetic vascular complications. Diabetes Care, 1996. 19(3): p. 257-267.
33. Curcio, F. and A. Ceriello, Decreased cultured endothelial cell proliferation in high glucose medium is reversed by antioxidants: New insights on the pathophysiological mechanisms of diabetic vascular complications. In Vitro Cellular and Developmental Biology, 1992. 28A: p. 787-790.
34. Natarajan, R., et al., Formation of an F2-isoprstane in vascular smooth muscle cells by elevated glucose and growth factors. American Journal of Physiology, 1996. 271(1): p. H159-H165.
35. Du, X.L., et al., Induction of apoptosis by high proinsulin and glucose in cultured human umbilical vein endothelial cells is mediated by reactive oxygen species. Diabetologia, 1998. 41: p. 249-256.
36. Kusuhara, M., et al., Oxidized LDL stimulates mitogen-activated protein kinases in smooth muscle cells and macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology, 1997. 17(1): p. 141-148.
37. Nofer, J.-R., et al., Low-density lipoproteins inhibit the Na+/H+ antiport in human platelets. Circulation, 1997. 95: p. 1370-1377.
38. Chappey, O., et al., Advanced glycation end products , oxidant stress and vascular lesions. European Journal of Clinical Investigation, 1997. 27: p. 97-108.
39. Pugliese, G., et al., Upregulation of mesangial growth factor and extracellular matrix synthesis by advanced glycation end products via a receptor-mediated mechanism. Diabetes, 1997. 46: p. 1881-1887.
40. Kunt, T., et al., The influence of advanced glycation endoproducts (AGE) on the expression of human endothelial adhesion molecules. Experimental and Clinical Endocrinology and Diabetes, 1998. 106(3): p. 183-138.
41. Cavalot, F., et al., Nonenzymatic glycation of fibronectin impairs adhesive and proliferative properties of human vascular smooth muscle cells. Metabolism, 1996. 45(3): p. 285-292.
42. Satoh, H., et al., Advanced glycation end products stimulate mitogen-activated protein kinase and proliferation in rabbit vascular smooth muscle cells. Biochemical and Biophysical Research Communications, 1997. 239: p. 111-115.
43. Berk, B.C., et al., Agonist-mediated changes in intracellular pH: role in vascular smooth muscle cell function. Journal of Cardiovascular Pharmacology, 1988. 12: p. S104-114.
44. Morishita, R., et al., Potential role of an endothelium-specific growth factor, heptocyte growth factor, on endothelial damage in diabetes. Diabetes, 1997. 46(1): p. 138-142.
45. Umeda, F., et al., Glucose reduces PDGF production and cell proliferation of cultured vascular endothelial cells. Hormone Metabolism Research, 1991. 23(6): p. 274-277.
46. Weimann, B.J., E. Lorch, and H.R. Baumgartner, High glucose concentrations do not influence replication and prostacyclin release of human endothelial cells. Diabetologia, 1984. 27(1): p. 62-3.
47. Santilli, S.M., et al., The effects of diabetes on the proliferation of aortic endothelial cells. Annuals of Vascular Surgery, 1992. 6(6): p. 503-510.
48. Hayashi, J.N., et al., Effects of glucose on migration, proliferation and tube formation by vascular endothelial cells. Virchows Archive B Cell Pathology, 1991. 60: p. 245-252.
49. Gade, P.V., et al., Nitric oxide mediates hyperglycemia-induced defective migration in cultured endothelial cells. Journal of Vascular Surgery, 1997. 26(2): p. 319-326.
50. Zerbini, G., et al., Activity and expression of the Na+/H+ exchanger in human endothelial cells cultured in high glucose. Diabetologia, 1995. 38: p. 782-791.
51. Cutaia, M.V., et al., Effect of hypoxic exposure on Na+/H+ antiporter activity, isoform expression, and localization in endothelial cells. American Journal of Physiology, 1998. 275(19): p. L442-L451.
52. Sayeski, P.P. and J.E. Kudlow, Glucose metabolism to glucosamine is necessary for glucose stimulation of transforming growth factor-alpha gene transcription. The Journal of Biological Chemistry, 1996. 271(25): p. 15237-15243.
53. Rumble, J.R., et al., Vascular hypertrophy in experimental diabetes: Role of advanced glycation products. Journal of Clinical Investigation, 1997. 99(5): p. 1016-1027.
54. Cagliero, E., et al., The effects of high glucose on human endothelial cell growth and gene expression are not mediated by transforming growth factor-beta. Laboratory Investigations, 1995. 73(5): p. 667-673.
55. Williams, B. and R.L. Howard, Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. American Society for Clinical Investigations, 1994. 93(6): p. 2623-2631.
56. Siczkowski, M. and L.L. NG, Glucose-induced changes in activity and phosphorylation of the Na+/H+ exchanger, NHE-1, in vascular myocytes from Wistar-Kyoto and Spontaneously Hypertensive Rats. Metabolism, 1996. 45(1): p. 114-119.
57. Sabri, A., et al., Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circulation Research, 1998. 82(10): p. 1053-1062.
| Discussion Board | Previous Page | Your Symposium |