Invited Symposium: What Can Genetic Models Tell Us About Attention-Deficit Hyperactivity Disorder (ADHD)?
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Attention deficit hyperactivity disorder (ADHD) is a major pediatric neuropsychiatric disorder affecting about 5% of school-aged children (1). The three major symptoms of inattention, excess impulsivity, and uncontrolled hyperactivity that define ADHD reflect a wide constellation of age-inappropriate behaviors which tend to cluster leading to the assignment of two subtypes of ADHD, inattention and hyperactivity/impulsivity. While affected children may independently present either subtype, the combined phenotype of inattention, impulsivity and hyperactivity appears to be most common. The behavioral impairments associated with ADHD are typically exhibited early in childhood, before the age of seven. Although the severity of these deficits, particularly hyperactivity, generally wane at adolescence, elements of these impairments do persist in 50-80% of adolescents and 30-50% of adults. Considerable evidence from family studies, and twin and adoption studies has strongly suggested that ADHD is heritable, although the penetrance is clearly not complete. Taken together these observations suggest that, as other complex neuropsychiatric disorders such as schizophrenia, ADHD is a multifaceted, likely heterogeneous disorder that arises from a variety of genetic interactions contributed by different gene loci. The genetic loci which contribute to the variable impairments of ADHD thus are likely to represent a collection of susceptibility or modulatory genes that initially influence development and consequently impact on information processing in the mature brain.
Despite compelling evidence for genetic heritance ADHD, few animal models of ADHD have emerged. However, characteristics of ADHD drawn from patient populations can be used to help identify certain elements of the disorder that may be modeled in animals, in particular rodents. For example, the observation that indirect dopamine agonists, particularly methylphenidate and d-amphetamine which effectively improve both deficits of hyperactivity and classroom performance, together with altered levels of dopamine metabolites provides support for the notion that dopaminergic pathways are particularly affected in ADHD. Morphometric studies based on magnetic resonance imaging have, in fact, provided additional evidence for the involvement of prefrontal cortex-striatal systems which receive strong dopaminergic input (2). Recently, Barkley has proposed that the principal deficit in ADHD lies in executive functions performed by the prefrontal cortex to control the initiation of motor responses leading to impulsivity and hyperactivity (3). Since prefrontal cortex receives modulatory input from the striatum, this again suggests a link between the circuitry of striatal and prefrontal dopaminergic pathways and behavioral deficits in ADHD. Studies examining the co-inheritance of polymorphic alleles of the dopamine reuptake transporter DAT1 and the dopamine D4 receptor DRD4 [reviewed in ref. 5] have identified specific alleles of these genes that may provide increased risk for ADHD. While these findings still appear controversial (4) and see ref. 5, they have provided specific gene targets to evaluate for a direct role in ADHD. It is of interest, therefore, that mice homozygous for the null DAT1 alleles, generated by homologous recombination gene "knock-out" technology, are spontaneously hyperactive. However, in the case of the DAT1 polymorphism associated with ADHD, this mutation lies outside the protein coding region and within the 3' untranslated portion of the mRNA. Thus it remains unclear how this might affect dopamine reuptake function in ADHD and correspond to the lack of dopamine transporter function in DAT1 knockout mice.
Several rat models displaying characteristics of ADHD have been described, including the genetic models based on the spontaneously hypertensive rat (SHR) (6) and Naples High-Excitability (NHE) (7) lines, as well as neonatal lesions of the dopaminergic input to the striatum (8). Behavioral and pharmacological studies have shown that such genetic and lesion models do faithfully reflect ADHD-associated deficits including, although not limited to, hyperkinesis (for example, see papers presented in this symposium by V.A. Russell; C.F. Deschepper et al.; F. Gonzalez-Lima and A.G. Sadile; and T. Sagvolden). However these models have been limited by the lack of precise mapping of genetic defects to trace the molecular mechanisms underlying the brain dysfunctions relevant to ADHD (but see the paper presented by M.P. Moisan, et al. for recent progress in this direction). In contrast, studies in the mouse have benefited enormously from the vast collection of genetic information and great number of genetically defined mutants, which when coupled with the advance of transgenic technology, and more recently targeted gene disruption methods, provide an effective means to identify genes that influence behavior. Based on this genetic groundwork, investigations in the mouse that complement the pharmacological and behavioral insight gained in rat models can begin to unravel the complexities of gene expression and development that underlie neuropsychiatric diseases, such as ADHD.
Results
The Coloboma Mutant Mouse
Coloboma mutant mice display a variety of behavioral, neurophysiological and developmental deficits, of which a subset may be compared with those presented by ADHD children. Although the Cm mutation is early embryonic lethal when homozygous (9), adult heterozygote mice (genotype, Cm/+) are quite viable but exhibit pronounced spontaneous hyperactive locomotor behavior, originally termed "circling," as well as head bobbing, and a distinctive ocular dysmorphology leading to sunken and often closed eyes (10). As described below, the hyperkinesis of these mutant mice averages three fold the activity of normal control littermates, but is quite variable suggesting a loss of control of activity rather than a simple increase in basal motor behavior. This hyperactivity can be effectively ameliorated by low doses of d-amphetamine, but not methylphenidate, resulting in normal levels of activity without induction of stereotypy (11). Evidence suggests that this hyperactive behavior is mediated by a deficiency in synaptic release of dopamine input to the striatum (12) that results from haploinsufficiency of the gene encoding SNAP-25, a key component of the synaptic vesicle docking and fusion complex required for regulated synaptic transmission. In addition to a loss of locomotor control, the deficiency in SNAP-25 expression seen in these mutants may also be linked to deficits in acquiring complex neurodevelopmental milestones in behavior (13), and in learning performance (Heyser, Wilson and Gold, in preparation) as well as hippocampal physiology (see below) that may correspond to impairments seen in ADHD.
The coloboma mutation is a chromosomal deletion that arose from neutron irradiation mutagenesis. Although neutron irradiation generally causes substantial chromosomal deletion and rearrangement, no chromosomal abnormality can be distinguished for the Cm locus at the cytological level (14). Initial mapping experiments placed the Cm locus on mouse chromosome 2 (15). Our more recent studies evaluating the inclusion of genetically mapped polymorphic simple sequence repeats and local gene sequences in an interspecies cross showed that the deletion spans 1-2 cM, comparable to 1-2 x 106 base pairs, towards the distal end of Ch2 (16). While a deletion of this sized segment of genomic DNA may comprise more than 30 genes, currently only four known genes (encoding phospholipase beta 1, and 4, jagged and SNAP-25 proteins) have been identified within the mutation (16, and M.C.W. unpublished data). The suggestion that a significant number of genes may be affected by the Cm deletion locus is consistent with the mutation being homozygous lethal. However while most null mutations, such as those produced in deletions, are recessive and result in little or no phenotypic change in heterozygotes, the Cm mutation appears semidominant resulting in a multifaceted phenotype in Cm/+ mice consistent with a contiguous gene defect.
 click to enlarge
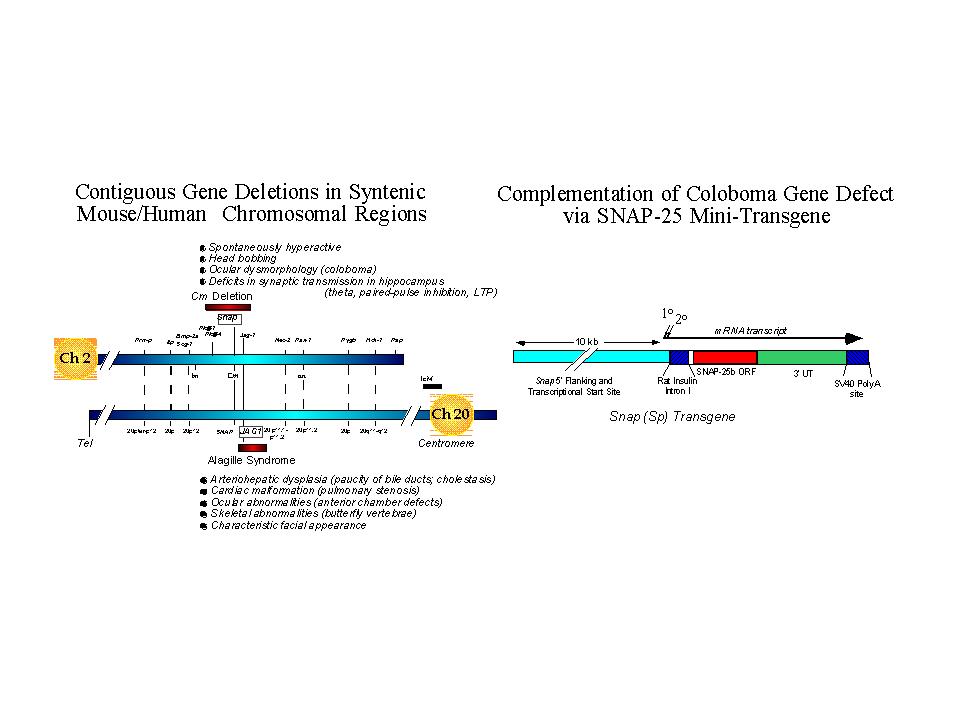
Fig. 1: Left Panel. Genetic maps of syntenic regions of mouse Ch 2 and human Ch 20 depicting relative positions of the coloboma and Alagilles' deletion loci, respectively. Positions of known genes are indicated with relative cytogenetic map positions for the human gene sequences. The major phenotypic abnormalities associated with each mutation are indicated. Whereas the hyperactivity produced by the coloboma mutation can be attributed to the gene Snap encoding SNAP-25, the critical gene for Alagilles' is JAG1 (see text for references).
Right Panel. A SNAP-25 minigene was constructed using promoter sequences 5' of the mouse gene sequence conferring neural specific expression and transcriptional start sites and a cDNA encoding the predominant SNAP-25b isoform expressed in adult brain. The rat insulin intron and SV40 polyadenylation sites were inserted for post-transcriptional processing and to promote effective expression. The expression of the transgene was assessed by in situ hybridization and found to be predominantly neuronal and consistent with the pattern of expression seen for the endogenous gene. (for additional details, see Hess, Collins and Wilson, 1996).
click to enlarge
Fig. 1: Left Panel. Genetic maps of syntenic regions of mouse Ch 2 and human Ch 20 depicting relative positions of the coloboma and Alagilles' deletion loci, respectively. Positions of known genes are indicated with relative cytogenetic map positions for the human gene sequences. The major phenotypic abnormalities associated with each mutation are indicated. Whereas the hyperactivity produced by the coloboma mutation can be attributed to the gene Snap encoding SNAP-25, the critical gene for Alagilles' is JAG1 (see text for references).
Right Panel. A SNAP-25 minigene was constructed using promoter sequences 5' of the mouse gene sequence conferring neural specific expression and transcriptional start sites and a cDNA encoding the predominant SNAP-25b isoform expressed in adult brain. The rat insulin intron and SV40 polyadenylation sites were inserted for post-transcriptional processing and to promote effective expression. The expression of the transgene was assessed by in situ hybridization and found to be predominantly neuronal and consistent with the pattern of expression seen for the endogenous gene. (for additional details, see Hess, Collins and Wilson, 1996).
As shown in the left panel of Figure 1, the gene encoding SNAP-25, Snap, lies approximately in the center of the Cm deletion and is flanked proximal by phospholipase genes Plcb-1, and b-4, and distal by Jag-1 on mouse chromosome 2. Interestingly, the semidominant herited human disorder Alagille's syndrome, characterized by arteriohepatic dysplasia, cardiac, ocular and skeletal abnormalities, is found on the syntenic region of human chromosome 20. While some patients exhibit contiguous gene deletions that can include the SNAP gene, the single obligatory gene for the core symptoms of Alagille's has been demonstrated to be JAG1 which encodes Jagged, a ligand for the master developmental regulator Notch (17, 18). Some Alagille's patients exhibit elements of mental retardation; however whether this correlates with a SNAP gene defect or if they manifest other deficits consistent with ADHD has not been resolved.
Deficiencies in SNAP-25 Lead to Dysregulation of Activity and Hyperkinesis
The role of SNAP-25 in mediating vesicular release of neurotransmitter suggested that it may be a candidate for neurophysiological dysfunction seen in Cm/+ mice. SNAP-25 (synaptosomal associated protein of 25kD) is a nerve terminal protein which together with syntaxin 1a and VAMP-2 (synaptobrevin-2) form a highly stable ternary complex (19). This complex is thought to form a direct link between transmitter laden synaptic vesicles and the plasma membrane at the site of fusion for exocytosis of neurotransmitter, for review see (20-22). In addition to tethering the vesicle to the active site of the presynaptic terminal, this core complex may serve as a molecular scaffold for assembly of other components required for Ca+2 triggered membrane fusion and recycling, such as synaptotagmin, NSF and (,(, and ( SNAPs (soluble NSF attachment proteins). Termed t- and v-SNAREs for their corresponding positions on vesicle and target plasma membranes, recent evidence has been presented suggesting that this complex of SNAP-25, syntaxin and VAMP may play a more direct role in triggering the fusion of these opposing membranes (23). The obligatory role of these proteins for neurotransmitter release and synaptic function is highlighted by their identification as specific substrates of botulinum neurotoxins, zinc-dependent proteases that cleave target proteins and completely block neuroexocytosis (24). Deficits in SNAP-25, therefore, would be expected to lead to a decrease in release of neurotransmitter, rather than a direct deficit in reuptake or receptor activation.
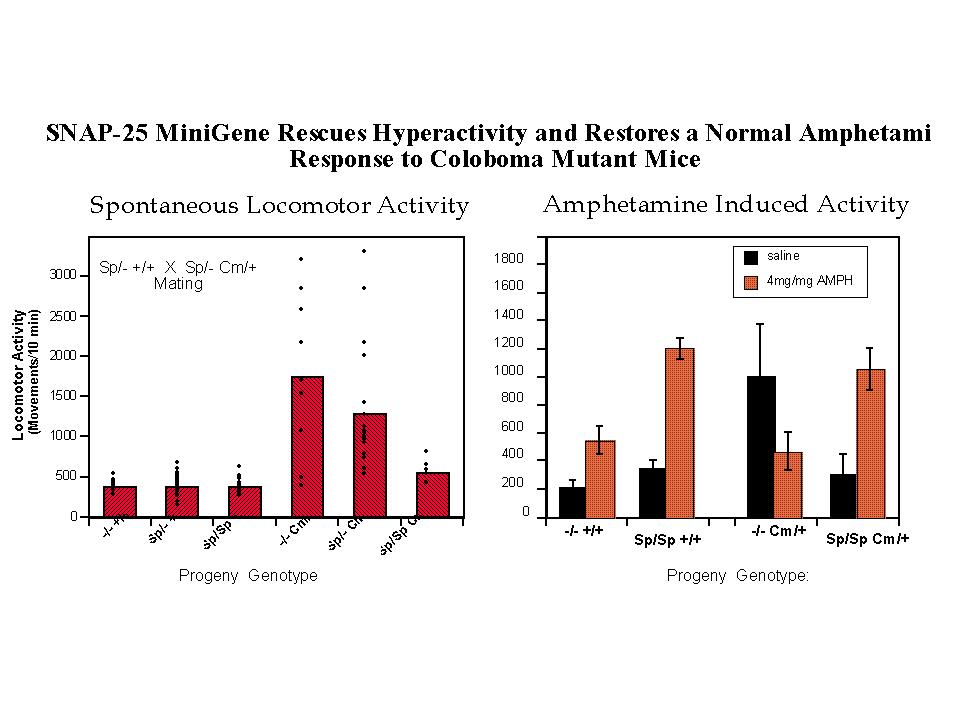
Coloboma mutant mice exhibit approximately 50% levels of SNAP-25 protein and mRNA (25), consistent with a dose dependent decrease resulting from the loss of one allele at the Cm deletion locus. To examine the contribution of a deficiency in SNAP-25 expression in the Cm mutant phenotype, a genetic complementation experiment was performed using transgenic mice carrying a SNAP-25 minigene (see Figure 1 and Hess et al. 1996 for details on construction of the mini-transgene and transgenic mice). Mating of mice heterozygous for the SNAP-25 transgene with mice both heterozygous for the transgene and Cm locus results in progeny of all potential allelic combinations (Figure 2, left). Importantly while the transgene had no apparent effect on wild-type progeny, mice bearing the Cm/+ mutation (identified by probing for Plcb-1), and homozygous for the SNAP-25 transgene were restored to normal activity. In addition, progeny heterozygous for both the transgene and Cm (genotype Sp/+ Cm/+) showed a trend for reduced locomotor activity consistent with a dose-response of the rescue by the transgene. Consistent with the restoration of normal activity, the SNAP-25 transgenes also restored a normal response to the psychostimulant d-amphetamine. As shown in Figure 2 (right panel) a moderately low dose of d-amphetamine (4 mg/kg, but similar results are seen with 2 mg/kg) induced a 2.6 fold increase of activity in wild-type mice whereas the activity of Cm/+ mutants was markedly decreased. In contrast, mice homozygous for the transgene and either bearing theCm locus or not showed a similar 3.5 fold induction in activity at this drug dose. While this provides strong evidence for the role of SNAP-25 in the hyperactive component of the coloboma phenotype, the frequency of the other characteristics of head bobbing or ocular dysmorphology were not affected by the SNAP-25 transgene (11), suggesting that these traits are determined by one or more distinct, though neighboring genes.
 click to enlarge
Fig. 2: Transgenic Rescue of Hyperactivity. Mice heterozygous for the SNAP-25 transgene (Sp) and one parent bearing the coloboma (Cm) mutation [parental genotypes: Sp/+ +/+ X Sp/+ Cm/+] were mated to provide offspring of all possible combinations of genotype. Progeny of this mating were tested for locomotor activity using a photobeam activated locomotor activity cage.
Left panel: Total locomotor activity was averaged over 3 hours in 10 minute bins (n=92 mice). Activity measurements of individual mice as well as the average of each genotype is shown.
Right panel: Locomotor activity induced by a low, submaximal dose of amphetamine (4mg/kg) was assessed for one hour in 10 minute bins starting 10 min after i.p. injection of the drug or saline carrier.
Note that the relative effect of amphetamine in wildtype mice (+/+) with or without Sp transgenes (3.4 and 2.6 fold increase, respectively) is similar to that shown by "transgenic rescued" Sp/Sp Cm/+ mice (3.5 fold increase), in contrast to coloboma mice lacking the transgene (-/- Cm/+) which exhibit a greater than two fold decrease in activity in response to the drug. For details, see Hess, Collins and Wilson, 1996.
click to enlarge
Fig. 2: Transgenic Rescue of Hyperactivity. Mice heterozygous for the SNAP-25 transgene (Sp) and one parent bearing the coloboma (Cm) mutation [parental genotypes: Sp/+ +/+ X Sp/+ Cm/+] were mated to provide offspring of all possible combinations of genotype. Progeny of this mating were tested for locomotor activity using a photobeam activated locomotor activity cage.
Left panel: Total locomotor activity was averaged over 3 hours in 10 minute bins (n=92 mice). Activity measurements of individual mice as well as the average of each genotype is shown.
Right panel: Locomotor activity induced by a low, submaximal dose of amphetamine (4mg/kg) was assessed for one hour in 10 minute bins starting 10 min after i.p. injection of the drug or saline carrier.
Note that the relative effect of amphetamine in wildtype mice (+/+) with or without Sp transgenes (3.4 and 2.6 fold increase, respectively) is similar to that shown by "transgenic rescued" Sp/Sp Cm/+ mice (3.5 fold increase), in contrast to coloboma mice lacking the transgene (-/- Cm/+) which exhibit a greater than two fold decrease in activity in response to the drug. For details, see Hess, Collins and Wilson, 1996.
Inspection of the activity of individual mice illustrates the point of dysregulation in behavioral control exhibited by these mutants. The extent of hyperkinesis of Cm/+ mice appears quite heterogeneous ranging from mice having quite normal locomotor activity to those reaching 10 fold greater activity over the extended period of the assay. It should be noted that these measures of activity (taken as a 10 min. averages over 3 hours) were made during the nocturnal or active phase of the mouse. The individuality of locomotor activity seen here is largely reproducible upon repeated testing of the same mouse (unpublished obs.) suggesting that the variability between mice may reflect individual control "set points" established by epigenetic factors during development. This differs markedly from the tight clustering and consequently highly regulated locomotor activity of wild-type mice (i.e. lacking the Cm locus) either with or without transgenes, and most importantly of homozygous transgene rescued mutants (Sp/Sp Cm/+). Interestingly, mice heterozygous for both transgene and Cm mutation showed a conspicuous clustering of lower activity values not represented in the noncomplemented -/- Cm/+ mice. Although this clustered activity is still two fold greater than normal, this might reflect the return of some, if not a complete, level of control exerted by the lower level of SNAP-25 transgene expression in these mice.
Hippocampal Electrophysiology
We examined synaptic function in coloboma mutant and normal mice by electroencephalograph (EEG) and evoked potential recordings in hippocampus of anesthetized mice (26). The advantage of this preparation is that the nervous system remains intact and can be examined for its response to external stimuli. The hippocampus, moreover, plays an important role in a variety of behaviors including learning and memory consolidation, but importantly also in the regulation of initiation of locomotor activity (27). While the precise function of dopamine as a neurotransmitter in the hippocampus remains unclear, anatomical evidence of dopaminergic projections from both the substantia nigra and ventral tegmental area, together with a number of pharmacological studies suggest that dopaminergic input can produce both direct and indirect modulatory effects on hippocampal physiology (for further discussion, see references in Steffensen, Wilson and Henriksen, 1996).
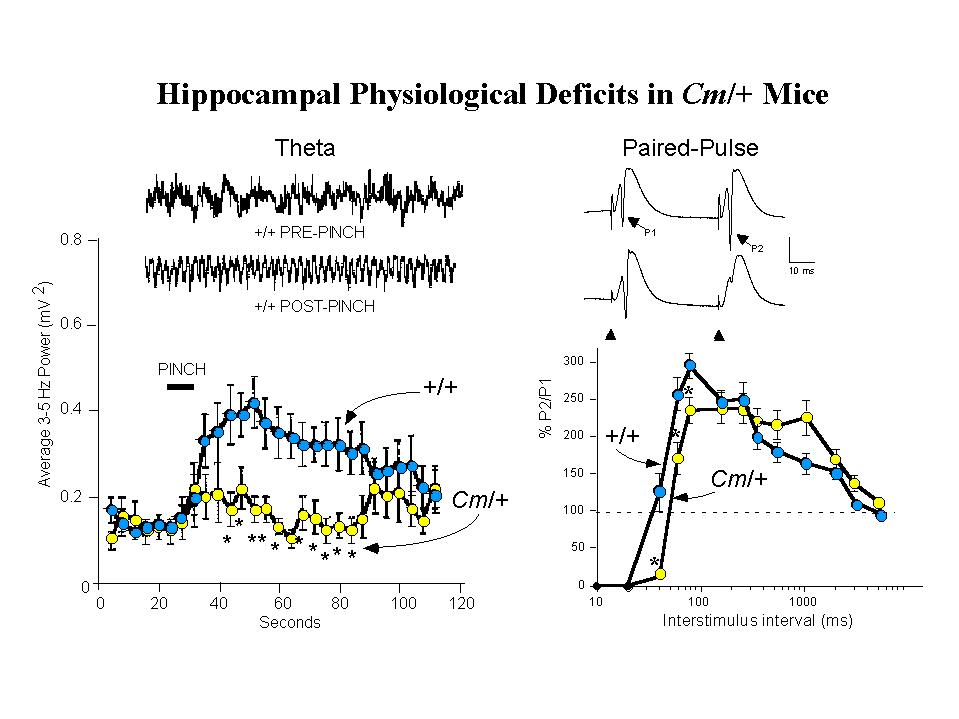
EEG recordings from the hilar region of the hippocampus were used to assess spontaneous and induced theta activity (3.0-5.0 Hz). Theta rhythm is a prominent waveform activity generated within the hippocampus, paced by rhythmically firing cells in the medial septum, that may underlie information processing in the hippocampus. Evidence suggests, moreover, that hippocampal theta associated with processing of motor information is modulated by dopamine (28). In anesthetized normal mice, application of a brief tail pinch provides an external stimuli that elicits a robust induction of synchronous theta activity. In contrast, coloboma mutant mice show a greatly diminished response to the same stimulus, although spontaneous theta activity seems unaltered (Figure 3, left panel). This suggests a disruption, possibly of dopaminergic input, in the circuitry mediating activation of theta in Cm/+ mutants (26). As seen for the genetic rescue of hyperactivity, examination of tail-pinch induced activation of theta in SNAP-25 transgenic rescued mutants demonstrated that this also was restored by the SNAP-25 transgene (Steffensen, Henriksen and Wilson, in preparation).
 click to enlarge
Fig. 3: Coloboma Mice Exhibit Deficits in Hippocampal Physiological Functions.
Left Panel: Tail Pinch Induced Hippocampal Theta Rhythm is Reduced. Insets show representative traces of hippocampal EEG activity recorded from the dentate gyrus of anesthetized wild-type (+/+) mice before and after a 10 sec tail-pinch stimulation. When compared with +/+ mice, the averaged 3-5 Hz EEG spectral power band of Cm/+ mice was decreased following tail-pinch).
Right Panel: Recurrent Inhibition is Increased in the Dentate Gyrus. Representative recordings from wild-type (+/+, upper trace) and coloboma (Cm/+, lower trace) are shown. The population spikes (PSs) evoked by perforant path stimulation after a 40 msec interval in +/+ mice are indicated (P1 and P2) in the upper trace., and the position of the stimulus artifact is indicated in the lower trace. In +/+ mice, paired-pulse potentiation of PSs occurred at interstimulus intervals of 40-2060 msec and absolute inhibition occurred at interstimulus intervals of less than 40 msec. In Cm/+ mice, in contrast, showed significantly increased inhibition at 40-80 msec intervals. For details, see Steffensen, Wilson and Henriksen 1996.
click to enlarge
Fig. 3: Coloboma Mice Exhibit Deficits in Hippocampal Physiological Functions.
Left Panel: Tail Pinch Induced Hippocampal Theta Rhythm is Reduced. Insets show representative traces of hippocampal EEG activity recorded from the dentate gyrus of anesthetized wild-type (+/+) mice before and after a 10 sec tail-pinch stimulation. When compared with +/+ mice, the averaged 3-5 Hz EEG spectral power band of Cm/+ mice was decreased following tail-pinch).
Right Panel: Recurrent Inhibition is Increased in the Dentate Gyrus. Representative recordings from wild-type (+/+, upper trace) and coloboma (Cm/+, lower trace) are shown. The population spikes (PSs) evoked by perforant path stimulation after a 40 msec interval in +/+ mice are indicated (P1 and P2) in the upper trace., and the position of the stimulus artifact is indicated in the lower trace. In +/+ mice, paired-pulse potentiation of PSs occurred at interstimulus intervals of 40-2060 msec and absolute inhibition occurred at interstimulus intervals of less than 40 msec. In Cm/+ mice, in contrast, showed significantly increased inhibition at 40-80 msec intervals. For details, see Steffensen, Wilson and Henriksen 1996.
Measurements of afferent-evoked population excitatory postsynaptic potentials (pEPSPs) in the perforant path have revealed additional abnormalities in hippocampal physiology ofCm/+ mutants (26). While the amplitude of EPSP facilitation appears unaltered in the mutants, analysis of population spikes elicited by paired-pulse stimulation showed that Cm/+ mice exhibit significantly increased recurrent inhibition over interstimulus intervals of 40-80 ms (Figure 3, right panel). Similar analysis has shown that in transgenic rescued Sp/Sp Cm/+ mice, paired-pulse inhibition of population spike amplitudes is returned to normal intervals of less than 40 ms (manuscript in preparation). In wild-type mice, d-amphetamine (3.0 mg/kg i.p.) produces significant disinhibition of paired-pulse responses which can be shown to be associated with spontaneous theta rhythm. In Cm/+ mice this dose of amphetamine, which restores normal locomotor activity to the mutants, conversely increases the inhibition of paired-pulse responses. This abnormal response to the indirect dopamine agonist amphetamine was also reversed by complementation by SNAP-25 transgenes (in preparation). These findings reinforce the idea that the impaired hippocampal physiology seen in coloboma mutant mice, as the apparent loss of control leading to hyperactivity, is mediated by deficits in dopaminergic synaptic transmission that can be attributed to deficiencies in SNAP-25 expression and function in neurotransmitter release.
Coloboma Mutants Exhibit Selective Deficits in Dopaminergic Transmission
Direct evidence for a disruption of dopaminergic transmission in Cm/+ mutant mice has been obtained using release assays from slice preparations in vitro (12). In this study, we compared calcium-dependent release of endogenous dopamine and serotonin (5-HT) from dorsal striatum, ventral striatum (including olfactory tubercle and nuc. accumbens) and cortex. In contrast to the significant KCl-induced release of dopamine from wild-type mice, no detectable depolarization evoked release of dopamine was observed from dorsal striatum of Cm /+ mutants. Interestingly, a small but significant increase in dopamine release was also found in cortex of Cm/+ mice, while the levels of evoked release from ventral striatum of mutant and wild-type mice were not significantly different. Moreover, the release of 5-HT from dorsal striatum of coloboma mice was diminished by about 50% compared with wild-type, although no differences were seen betweenCm/+ and +/+ mice in cortex and ventral striatum. In contrast the marked differences observed for KCl-induced release in dorsal striatum of mutant and wild-type mice, no difference was detected for the basal level of dopamine or 5-HT.
Discussion and Conclusion
Our studies suggest that the deficiency of SNAP-25 expression exhibited in coloboma mutant mice contributes to a loss of control of locomotor activity leading to hyperactivity, and deficits in hippocampal function which may underlie impairments to information processing. The selective effect on depolarization evoked release of dopamine and 5-HT is consistent with a defect in neuroexocytosis known to be dependent on SNAP-25. Given this observation, it is likely that amphetamine which promotes non-vesicular release of dopamine through the reversal of the plasma membrane transporter (29) restores control and reduces the hyperactivity of Cm/+ mice because of its ability to circumvent a block in vesicular release caused by the deficiency of SNAP-25 on nigra-striatal terminals. Consequently, methylphenidate which specifically blocks reuptake and appears to be unable to reverse the hyperactivity of coloboma mice at the doses tested (11) may be comparatively ineffective in raising synaptic dopamine at SNAP-25 deficient terminals to sufficient levels to restore motor control.
The finding that regulated release of dopamine and 5-HT is prominently affected in dorsal striatum, but not ventral striatum indicates the possibility that synaptic function in specific brain regions may selectively be vulnerable to defects in presynaptic mechanisms of neurotransmitter release. Whether this mechanism, seen in coloboma mutant mice, corresponds to the structural and functional deficits in striatal input and prefrontal cortical regulation of executive functions controlling behavior that have been proposed for ADHD (see ref. 2 and 3) remains to shown. It is interesting to note in this respect that although both d-amphetamine and methylphenidate have been proven to be effective therapeutics for improving behavioral and classroom performance, there is considerable individual variability in clinical responses to these drugs (see ref. 30). This suggests the possibility that, at least in some cases, deficits in presynaptic release, similar to that found in the coloboma mutant mouse, may contribute to the behavioral and neuropsychiatric dysregulation typical of ADHD. While it remains unclear whether alterations in SNAP-25 gene expression do directly play a role as one of the multiple genetic factors contributing to ADHD (31), it is clear that the coloboma mouse mutant, and specifically SNAP-25 deficient mice, will provide a well-defined model to explore the impact of dysfunction of synaptic transmission mechanics in behavioral and neuropsychiatric disease.
References
1. Szatmari, P. (1992) Child. Adolesc. Psychiatr. Clin. North Am. 1, 361-371.
2. Castellanos, F. X., Giedd, J. N., W.L., M., Hamburger, S. D., Vaituzis, A. C., Dickstein, D. P., Sarfatti, S. E., Vaus, Y. C., Snell, J. W., Rajapakse, J. C. & Rapoport, J. L. (1996) Arch. Gen. Psych. 53, 607-616.
3. Barkley, R. A. (1997) Dev. Behav. Pediatrics 18, 271-279.
4. Castellanos, F. X., Lau, E., Tayebi, N., Lee, P., Long, R. E., Giedd, J. N., Sharp, W., Marsh, W. L., Walter, J. M., Hamburger, S. D., Ginns, E. I., Rapoport, J. L. & Sidransky, E. (1998) Mol. Psychiatry 3, 431-434.
5. Tharpar, A. (1998) Mol. Psychiatry 3, 370-372.
6. Sagvolden, T., Pettersen, M. B. & Larsen, M. C. (1993) Physiol. Behav. 54, 1047-1055.
7. Cerbone, A., Patacchioli, F. R. & Sadile, A. G. (1993) Behav. Brain Res. 55, 1-16.
8. Kostrzewa, R. M., Brus, R., Kalbfleisch, J. H., Perry, K. W. & Fuller, R. W. (1994) Brain Res. Bull. 34, 161-167.
9. Theiler, K. & Varnum, D. S. (1981) Anat. Embryol. 162, 121-126.
10. Searle, A. G. (1966) Mouse News Lett. 35, 27.
11. Hess, E. J., Collins, K. A. & Wilson, M. C. (1996) J. Neurosci. 16, 3104-3111.
12. Raber, J., Mehta, P. P., Kreifeldt, M., Parsons, L. H., Weiss, F., Bloom, F. E. & Wilson, M. C. (1997) J. Neurochem. 68, 176-186.
13. Heyser, C. J., Wilson, M. C. & Gold, L. H. (1995) Dev. Brain Res. 89, 264-269.
14. Davisson, M. T. & Lewis, S. E. (1990) in Branbury Report, ed. Allen, J. W. (Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.), Vol. 34, pp. 195-206.
15. Snell, G. D. & Bunker, H. P. (1967) Mouse News Lett. 37, 34.
16. Hess, E. J., Collins, K. A., Copeland, N. G., Jenkins, N. A. & Wilson, M. C. (1994) Genomics 21, 257-261.
17. Oda, T., Elkahloun, A. G., Pike, B. L., Okajima, K., Krantz, I. D., Genin, A., Piccoli, D. A., Meltzer, P. S., Spinner, N. B., Collins, F. S. & Chandrasekharappa, S. C. (1997) Nat Genet 16, 235-234.
18. Li, L., Krantz, I. D., Deng, Y., Genin, A., Banta, A. B., Collins, C. C., Qi, M., Trask, B. J., Kuo, W. L., Cochran, J., Costa, T., Pierpont, M. E., Rand, E. B., Piccoli, D. A., Hood, L. & Spinner, N. B. (1997) Nat. Genet. 16, 243-251.
19. Chapman, E. R., An, S., Barton, N. & Jahn, R. (1994) J. Biol. Chem. 269, 27427-27432.
20. Südhof, T. C. (1995) Nature 375, 645-653.
21. Calakos, N. & Scheller, R. H. (1996) Physiological Reviews 76, 1-29.
22. Bark, I. C. & Wilson, M. C. (1994) Proc. Natl. Acad. Sci. USA 91, 4621-4624.
23. Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., Sollner, T. H. & Rothman, J. E. (1998) Cell 92, 759-72.
24. Montecucco, C. & Schiavo, G. (1994) Mol. Microbiol. 13, 1-8.
25. Hess, E. J., Jinnah, H. A., Kozak, C. A. & Wilson, M. C. (1992) J. Neurosci. 12, 2865-2874.
26. Steffensen, S. C., Wilson, M. C. & Henriksen, S. J. (1996) Synapse 22, 281-289.
27. Morgenson, G. J. (1987) Prog. Psychobiol. 12, 117-170.
28. Henriksen, S. J., Criado, J. R. & Steffensen, S. C. (1994) Soc. Neurosci. Abst. 20, 801.
29. Sulzer, D., Chen, T.-K., Lau, Y. Y., Kristensen, H., Rayport, S. & Ewing, A. (1995) J. Neurosci. 15, 4102-4108.
30. Elia, J., Borcherding, B. G., Rapoport, J. L. & Keysor, C. S. (1991) Psychiat. Res. 36, 141-155.
31. Hess, E. J., Rogan, P. K., Domoto, M., Tinker, D. E., Ladda, R. L. & Ramer, J. C. (1995) Am. J. Med. Genet. 18, 573-579.
| Discussion Board | Previous Page | Your Symposium |