Poster Contents
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
Excitatory amino acids (EAA), glutamate and aspartate, are endogenous compounds acting as neurotransmitters in brain, through the activation of three types of ionotropic receptors: the NMDA (N-methyl-D-aspartate), the AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid) and the KA (kainic acid) receptors. NMDA receptor-associated channels are permeable to Na+, K+ and Ca2+ in a voltage-dependent manner whereas AMPA and KA receptors are linked to Na+ permeable channels. Finally, glutamate also activates metabotropic receptors which induce G protein-mediated events. However, excessive stimulation of these receptors by EAA, such as during cerebral ischemia, leads to neuronal degeneration. The injury can be separated into two components: an acute Na+/Cl--dependent neuronal swelling and a delayed Ca2+-mediated cell death (Choi, 1992). This latter phenomenon may involve both nitric oxide (.NO) and reactive oxygen species (ROS), i.e. superoxide anion radical (O2.-), hydrogen peroxide (H2O2) and their highly cytotoxic by-product hydroxyl radical (.OH).
.NO was shown to participate in glutamate injury in neuronal cultures (Dawson et al., 1991) and in tissue infarction after focal cerebral ischemia in rats (Buisson et al., 1992a; Iadecola et al., 1994). On the other hand, evidence has accumulated showing a formation of ROS under excitotoxic or ischemic conditions. Indeed, a production of O2.- was reported in neuronal cultures in the presence of NMDA (Lafon-Cazal et al., 1993). An .OH generation also occurred in vivo in rat striatum under NMDA (Hammer et al., 1993) or glutamate (Lancelot et al., 1995) exposure, and during focal or global cerebral ischemia (Ginsberg et al., 1994). However, there is as yet only limited evidence linking excitotoxicity to the generation of free radicals in vivo. Yet, we have previously shown that the outcome of an excitotoxic insult depended mainly on hydroxyl radicals produced during the postexcitotoxic period rather during the period of exposure to the excitotoxin (Lancelot et al., 1997).
In the present study, the microdialysis technique was used to perfuse glutamate into rat striatum. In this model of excitotoxicity, we examined the contribution of NMDA receptors to .OH generation and tissue injury.
Materials and Methods
General procedures
Sixty-six male Sprague-Dawley rats (300-350 g) were anaesthetized with chloral hydrate (400 mg/kg, i.p.). A microdialysis probe, prepared according to the technique described by Robinson and Whishaw (1988), with a 4 mm long dialysis membrane, was implanted vertically into the left dorsolateral striatum. Stereotaxic coordinates were as follows: 0.0 mm anterior and 3.5 mm lateral to the bregma, 7.0 mm ventral to the skull surface (according to the atlas of Paxinos and Watson, 1982). The probe was perfused with Ringer's solution (in mM: NaCl, 125; KCl, 2.5; MgCl2, 1.18; CaCl2, 1.26) at a constant flow rate (1.5 �l/min) before switching to experimental drug solutions. The experiment was performed in strict accordance with NIH guidelines.
Hydroxyl radicals were measured on awake animals by using the procedure described by Obata and Chiueh (1992). Briefly, the technique is based on the hydroxylation of sodium salicylate by .OH, leading to the production of 2,3- and 2,5-dihydroxybenzoic acids (2,3- and 2,5-DHBA). However, in the whole study, as salicylate is catabolized into 2,5-DHBA not only by .OH but also through the activation of mitochondrial P450 enzymes (Halliwell et al., 1991), we have only reported changes in 2,3-DHBA efflux. Therefore, after a 90 min equilibrium period, the perfusion medium was switched for 2 h to a Ringer's solution containing 5 mM sodium salicylate in order to define basal 2,3-DHBA levels. The effect of EAA on .OH production was then tested by adding either 500 mM sodium glutamate or 10 mM NMDA to the perfusion medium for another 2 h. Brain dialysates were collected every 30 min and assayed for 2,3-DHBA by an HPLC procedure with electrochemical detection. Each sample was injected via a 20 �l loop onto a 5 �m C18, 250 x 4 mm LiChrospher column. The mobile phase consisted of 30 mM sodium citrate, 30 mM sodium acetate and 4% methanol. The flow rate was 1.2 ml/min. The compounds were detected with the electrode set at +0.6 V against an Ag/AgCl2 reference electrode.
In order to quantify the size of the necrosis around the microdialysis probe, the rats were sacrificed 24 h after the perfusion. The brains were removed and frozen in isopentane (at -40�C). Coronal sections (50 �m) were cut on a cryostat microtome every 500 �m, starting at 11.2 mm and continuing through 6.7 mm anterior to the interaural line (Paxinos and Watson, 1982). The sections were stained with cresyl violet. Then, the necrotic areas were measured using an image analyser. The extent of neuronal damage was assessed by the volume of the striatal lesion, which was determined by integration of the necrotic areas with the distance between each level.
Experimental protocol
Dizocilpine (MK-801) or 2-amino-5-phosphonopentanoic acid (AP-5) were used to ensure the blockade of NMDA receptors. The experimental groups were defined as follows:
Experiment 1:
Group I (n = 12): 2 h perfusion in basal conditions, followed by 2 h perfusion of glutamate.
Group II (n = 10): idem group I with co-perfusion of 100 �M MK-801 during the 4 h.
Group III (n = 9): idem group I with co-perfusion of 300 �M AP-5 during the 4 h.
Experiment 2:
Group I (n = 11): 2 h perfusion in basal conditions, followed by 2 h perfusion of glutamate.
Group II (n = 10): idem group I with i.p. injection of 5 mg/kg MK-801 30 min before glutamate perfusion.
Experiment 3:
Group I (n = 8): 2 h perfusion in basal conditions, followed by 2 h perfusion of NMDA.
Group II (n = 6): idem group I with i.p. injection of 5 mg/kg MK-801 30 min before glutamate perfusion.
Salicylate was purchased from Merck, MK-801 and AP-5 from RBI, glutamate and NMDA from Sigma Chemicals.
Data analysis
The concentrations of 2,3-DHBA measured in the dialysates were expressed in nM (means � SEM) whereas the total amounts of 2,3-DHBA generated during the 2 h perfusion of EAA were expressed in pmol (means � SEM). The volumes of necrosis were expressed in mm3 (means � SEM). The concentrations were analyzed by repeated measures ANOVA followed by Scheff� tests as a post-hoc procedure. For the amounts of 2,3-DHBA and the volumes of necrosis, multiple groups were evaluated by an ANOVA followed by a Scheff� test as a post-hoc procedure while two-group comparisons were made by the unpaired Student's t test. Differences were considered statistically significant at P<0.05. Linear regression analysis was used to show correlation between 2,3-DHBA amounts and necrosis volumes.
Results
In control animals in experiment I, after the first dialysate fraction, 2,3-DHBA concentrations reached a steady state and remained in a range of 10-30 nM (Fig. 1). Glutamate perfusion produced a marked increase in 2,3-DHBA output (x10 versus baseline, P<0.001), which peaked at 206 � 15 nM in the first dialysate fraction under glutamate exposure and remained near this value up to the end of the experiment. The addition of 100 �M MK-801 or 300 �M AP-5 into the perfusion medium reduced significantly this glutamate-induced 2,3-DHBA overflow (Fig. 1).
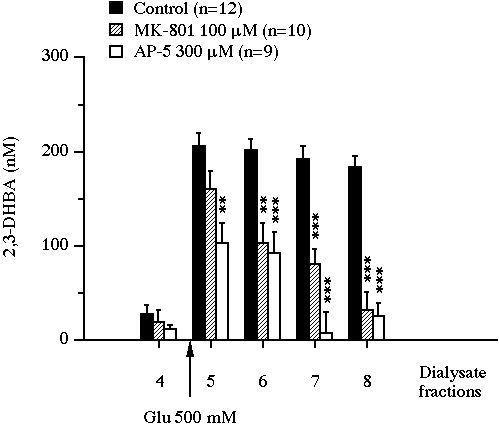
Fig 1 : Effect of the intrastriatal perfusion of two NMDA receptor antagonists on the evolution of 2,3-DHBA concentrations in microdialysis perfusates of rat striatum exposed to glutamate. Values were expressed as mean � SEM. ** P<0.01, *** P<0.001, compared with the value of control animals.
The cumulative amount of 2,3-DHBA collected during the 2 h glutamate perfusion was actually reduced from 35.3 � 1.5 pmol to 16.9 � 2.6 pmol (-52% versus control, P<0.001) by MK-801 and to 10.4 � 1.9 pmol (-71% versus control, P<0.001) by AP-5 (Fig. 2A). In control animals, the perfusion of glutamate led, after 24 h, to a striatal necrosis of 22.9 � 0.8 mm3 (Fig. 2B). MK-801 or AP-5 added to the perfusion medium were unable to modify the size of the glutamate-induced lesion (the volumes of necrosis were 22.5 � 0.8 mm3 and 23.1 � 0.8 mm3 respectively).
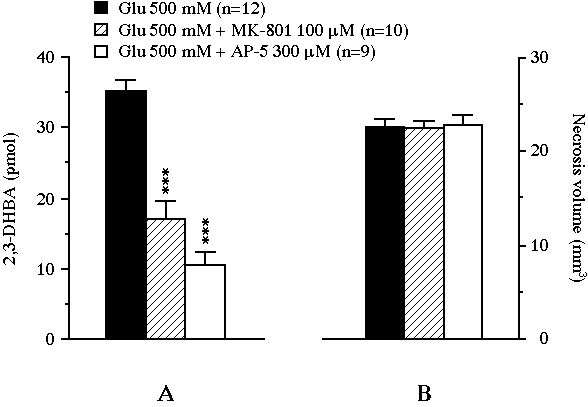
Fig 2 : Effect of the intrastriatal perfusion of two NMDA receptor antagonists on the total amount of 2,3-DHBA collected during the 2 h exposure of the striatum to glutamate (pannel A), and on the volume of the glutamate-induced necrosis (pannel B). Values were expressed as mean � SEM. *** P<0.001, compared with the value of control animals perfused with glutamate alone.
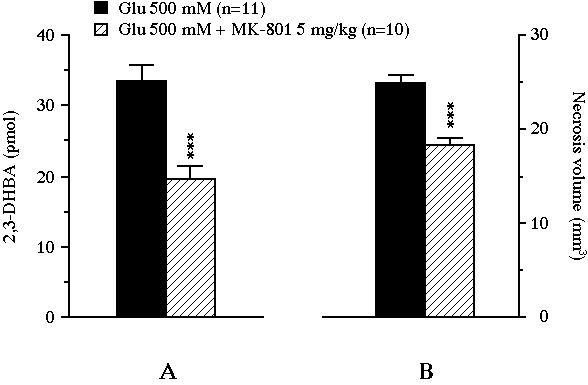
Fig 3 : Effect of an intraperitoneally administered NMDA receptor antagonist on the total amount of 2,3-DHBA collected during the 2 h exposure of the striatum to glutamate (pannel A), and on the volume of the glutamate-induced necrosis (pannel B). Values were expressed as mean � SEM. *** P<0.001, compared with the value of control animals perfused with glutamate alone.
In experiment II, the intraperitoneal administration of 5 mg/kg MK-801 resulted in a significant reduction of the glutamate-induced 2,3-DHBA overflow from 33.5 � 2.3 pmol to 19.6 � 1.9 pmol (-41%, P<0.001, Fig. 3A). This treatment also decreased the volume of the lesion from 24.9 � 0.8 mm3 to 18.2 � 0.9 mm3 (-27%, P<0.001, Fig. 3B). Moreover, the volume of necrosis correlated positively with the total amount of 2,3-DHBA formed during the 2 h glutamate perfusion (r = 0.46, n = 21, P<0.05, Fig. 4).
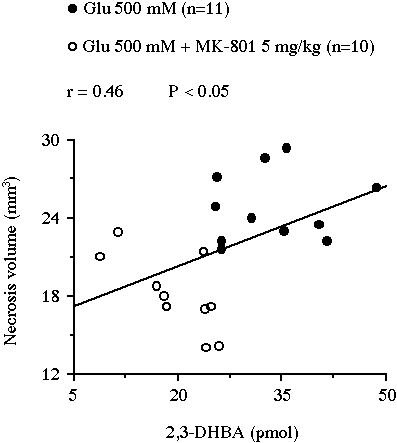 Fig 4 : Correlation between the volume of the striatal necrosis induced by a perfusion of glutamate and the total amount of 2,3-DHBA collected during the 2 h perfusion.
Fig 4 : Correlation between the volume of the striatal necrosis induced by a perfusion of glutamate and the total amount of 2,3-DHBA collected during the 2 h perfusion.
In experiment III, the perfusion of NMDA yielded a massive .OH production (x30 versus baseline, P<0.001) which peaked at 620 � 64 nM in the second dialysate fraction under NMDA exposure and remained elevated up to the end of the experiment (data not shown). This NMDA-induced 2,3-DHBA output was significantly lowered by the systemic administration of MK-801 (5 mg/kg, i.p.). Indeed, the cumulative amount of 2,3-DHBA generated during the 2 h NMDA perfusion was significantly decreased by MK-801 from 93.2 � 5.0 pmol to 66.2 � 12.4 pmol (-29%, P<0.05, Fig. 5A). The treatment also reduced the volume of the lesion from 11.4 � 0.8 mm3 in the control group to 4.3 � 0.5 mm3 in the MK-801-treated group (-63%, P<0.001, Fig. 5B). In this experiment, the volume of necrosis nearly correlated with the amount of 2,3-DHBA formed during the 2 h NMDA perfusion (r = 0.51, n = 14, P = 0.06, Fig 6).
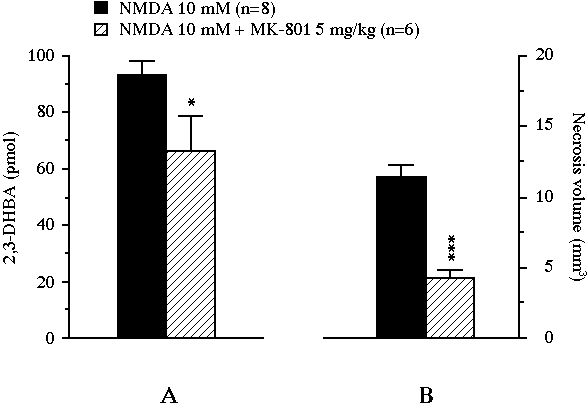
Fig 5 : Effect of an intraperitoneally administered NMDA receptor antagonist on the total amount of 2,3-DHBA collected during the 2 h exposure of the striatum to NMDA (pannel A), and on the volume of the NMDA-induced necrosis (pannel B). Values were expressed as mean � SEM. * P<0.05, *** P<0.001, compared with the value of control animals perfused with NMDA alone.
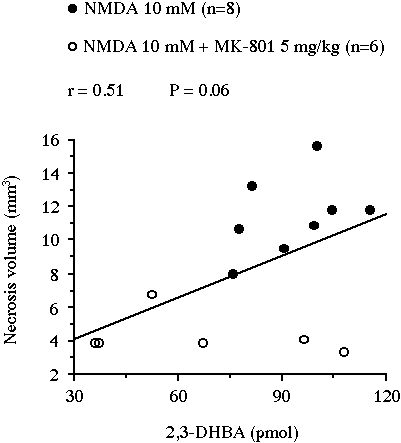 Fig 6 : Correlation between the volume of the striatal necrosis induced by a perfusion of NMDA and the total amount of 2,3-DHBA collected during the 2 h perfusion.
Fig 6 : Correlation between the volume of the striatal necrosis induced by a perfusion of NMDA and the total amount of 2,3-DHBA collected during the 2 h perfusion.
Discussion and Conclusion
An increasing amount of evidence has led authors to propose free radical production as a potential mechanism of brain injury in neurodegenerative disorders, stroke, and trauma. An .OH generation was actually reported in models of Parkinson�s disease (Obata and Chiueh, 1992), cerebral ischemia (Phillis and Sen, 1993; Ginsberg et al., 1994) and brain trauma (Hall et al., 1993). In these neurologic illnesses, increased extracellular EAA concentrations might be the cause of this ROS formation leading to oxidative stress and subsequent neuronal loss. In fact, evidence has accumulated that, when neurones are directly exposed to EAA, a production of ROS occurs possibly through mechanisms involving phospholipase A2, nitric oxide synthase or xanthine oxidase. For instance, a O2.- generation was reported in neuronal cultures exposed to glutamate or NMDA (Lafon-Cazal et al., 1993). Mepacrine, a phospholipase A2 inhibitor, could prevent this production. Therefore, pol yunsaturated fatty acids released from membranes by this enzyme have been considered as potential precursors of ROS in vivo. On the other hand, NG-nitro-L-arginine methyl ester, a nitric oxide synthase inhibitor, could reduce the efflux of .OH in rat striatum submitted to an NMDA perfusion (Hammer et al., 1993). It was suggested that, when .NO is produced in the presence of O2.-, both compounds can interact and form a peroxynitrite anion intermediate (ONOO-). As it was demonstrated in vitro (Beckman et al., 1990), this compound might decompose into .OH and nitrogen dioxide radical (.NO2) in vivo. In addition, the xanthine oxidase inhibitor oxypurinol virtually abolished the formation of oxyradicals in a rat model of global cerebral ischemia (Phillis and Sen, 1993). However, xanthine oxidase inhibition did not prevent .OH production when glutamate was perfused into rat striatum (Boisvert, 1992).
In the present study, a striatal excitotoxic lesion was produced by perfusing glutamate through a microdialysis probe, as previously described by Boisvert (1992). This model had the advantage compared to striatal injections of EAA of allowing at the same time an evaluation of the biochemical consequences of glutamate perfusion. Therefore, using the salicylate trapping technique developed by Obata and Chiueh (1992), we could reliably estimate .OH production before assessing the extent of the injury. Glutamate was perfused at the same concentration (500 mM) as it was previously used in this model (Boisvert, 1992; Lancelot et al., 1995). Considering that only 10% of the glutamate molecules present in the perfusate could cross the dialysis membrane, the concentration to which neurones were actually exposed (50 mM) remained in the range of concentrations used by other investigators to induce a striatal excitotoxic lesion: 240 mM quinolinate, an NMDA receptor agonist (Beal et al ., 1988), 300 mM NMDA (Chapman et al., 1989), or 200 mM NMDA (Schulz et al., 1995).
In the present study, by determining 2,3-DHBA levels, we confirmed our already published results describing an .OH production in rat striatum during a 500 mM glutamate perfusion (Lancelot et al., 1995). 2,5-DHBA concentrations increased similarly as 2,3-DHBA (data not shown) and were consistent with the data obtained by Boisvert (1992). Furthermore, when 500 mM glutamate was replaced by 10 mM NMDA in the perfusate, 2,3-DHBA levels were also massively increased (x30 versus baseline). However, Hammer et al. (1993) only reported a much moderate increase (x4 versus baseline) with the same NMDA concentration. This discrepancy may be explained by their basal 2,3-DHBA output of nearly 18 pmol /2 h, an approximately 6 fold higher amount than in the present study. Taken together, these results suggest that glutamate may yield .OH in vivo, at least via the activation of NMDA receptors, since NMDA itself produces .OH.
Histological examination of control groups in experiments I and II demonstrated that glutamate induces a reproducible striatal lesion. We have previously observed that the size of the necrosis is not modified by the presence of salicylate (data not shown). The neurotoxicity observed in the present study is consistent with the dramatic loss of neurons described by Boisvert and Schreiber (1992) in the striatum of rats 24 h after a 2 h perfusion with 250 mM glutamate. Moreover, the perfusion of NMDA also induced a necrosis, as it has been previously described after a direct injection of NMDA into the striatum (Buisson et al., 1992b). Subsequently, as they are in accordance with other experiments, our data demonstrate that the administration of EAA through the microdialysis probe is a convenient way for inducing an excitotoxic lesion which can be pharmacologically modulated and studied both biochemically and histologically.
We have shown here that the intrastriatal perfusion of either 100 �M MK-801 or 300 �M AP-5 reduces significantly the glutamate-induced production of .OH by 52 and 71% respectively. Similarly, the intraperitoneal administration of 5 mg/kg MK-801 decreased by 41 and 29% the .OH formation induced by glutamate or NMDA respectively. These results provide evidence of the involvement of NMDA receptors in the generation of ROS by neurons under glutamate exposure. They are in accordance with the study of Hammer et al. (1993) who reduced to baseline the NMDA-induced 2,3-DHBA overflow by adding 100 �M MK-801 to the perfusion medium. Moreover, our results are also supported by those of Lafon-Cazal et al. (1993) who completely abolished the NMDA-induced O2.- production in neuronal cultures by the use of either MK-801 or AP-5.
In spite of a reduced .OH formation, the volume of the lesion under glutamate exposure was not modified by the perfusion of 100 �M MK-801 or 300 �M AP-5. This unexpected result led us to envisage, first, that NMDA receptors may not be responsible for the excitotoxic lesion caused by glutamate in our experimental conditions and, second, that .OH may not be involved in this process. This latter hypothesis was corroborated by previous studies demonstrating that the neurotoxicity of quinolinate, an NMDA receptor agonist, was not prevented by the antioxidants ascorbic acid, beta-carotene and alpha-tocopherol (Beal et al., 1988). On the other hand, Beal et al. (1988) also reported that the striatal lesion caused by quinolinate is significantly reduced by an equimolar co-injection of AP-5 or by an intraperitoneal administration of 5 mg/kg MK-801. Therefore, we have tested the effect of a pre-treatment with MK-801 (5 mg/kg, i.p.) and, this time, we have found that the d rug is neuroprotective against both glutamate and NMDA neurotoxicity. This result demonstrates that, whereas the local application of an NMDA receptor antagonist does not reduce the extent of the lesion, this same drug is neuroprotective when given systemically. To explain this discrepancy, we propose that, once compounds perfused through the microdialysis probe have crossed the dialysis membrane, their concentrations rapidly decrease with distance to the probe according to a gradient. Moreover, it is very probable that glutamate and NMDA are cytotoxic at distances out of their diffusion area since they may spread their excitotoxic effect to neurons localized in more peripheral regions which, in turn, also release endogenous EAA. Subsequently, it is conceivable that tissues are not protected by locally applied NMDA receptor antagonists because the concentrations of these drugs are not sufficient in the outer areas of the lesion. On the contrary, the systemic administration of MK-801 can reduce the necrosis vo lume because the drug is able to reach active concentrations in these outlying parts of the lesion, where the extracellular concentrations of EAA are not too high.
As for the involvement of .OH in the injury caused by EAA, we provided here evidence that their production is correlated with the volume of necrosis induced by glutamate. Moreover, it was very nearly so with the NMDA perfusion. If we refer to the three conditions defined by Coyle and Puttfarcken (1993) which have to be fulfilled to enable distinguishing between oxidative stress as an epiphenomenon and a causal event, two at least of these criteria were satisfied in our study. Indeed, there was evidence of oxidative damage (i.e. membrane lipid peroxidation, proteolysis and DNA disruption) and the intraperitoneal administration of MK-801 reduced both .OH formation and neuronal degeneration. However, the perfusion of 10 mM NMDA induced a 2.7 times higher .OH production than 500 mM glutamate whereas the necrosis volume was twice smaller. Therefore, we cannot assert that NMDA receptor-mediated .OH are really involved in the glutamate-induced striatal lesion. W e must keep in mind, however, that we did not evaluate the overall exposure of the striatum to oxyradicals since .OH were only recorded acutely whereas the lesion was evaluated 24 h later. Yet, we have demonstrated previously that hydroxyl radical formation during the postexcitotoxic period is essential in the neurodegeneration of striatum exposed to glutamate (Lancelot et al., 1997)
In summary, the present study demonstrates that an intrastriatal perfusion of glutamate yields both a massive .OH generation and a substantial tissue damage, two phenomenons involving NMDA receptors. However, although the necrosis volume correlated positively with the amount of .OH produced during the perfusion of glutamate, we could not establish a causal relationship between .OH and injury in our experimental conditions. Moreover, NMDA receptors are probably not the only receptors responsible for the glutamate-induced oxidative stress and neurodegeneration. Sun et al. (1992) actually detected free radicals and observed an increased lipid peroxidation in gerbil brains 24 h after systemic injection of KA. Furthermore, the neurotoxic effects of intracerebrally administered KA or quisqualate, an AMPA receptor agonist, were blocked by the centrally active antioxidant, idebenone, which does not affect NMDA receptor-mediated neurotoxicity (Miyamoto and Coyle, 1990). Taken together, these results suggest that, beside NMDA receptors, KA and AMPA receptors as well may participate in the glutamate-induced neurodegeneration by mediating cytotoxic ROS formation. Therefore, while elaborating new therapeutic strategies aimed at protecting neuronal tissues in cerebral ischemia and other neurological disorders by suppressing the oxidative stress, the importance of these various sources of ROS should be kept in mind.
References
- Beal M. F., Kowall N. W., Swartz K. J., Ferrante R. J., and Martin J. B. (1988) Systemic approaches to modifying quinolinic acid striatal lesions in rats. J. Neurosci. 8, 3901-3908.
- Beckman J. S., Beckman T. W., Chen J., Marshall P. A., and Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 87, 1620-1624.
- Boisvert D. P. (1992) In vivo generation of hydroxyl radicals during glutamate exposure, in The role of neurotransmitters in brain injury (Globus M. and Dietrich W. D., eds), pp. 361-366. Plenum Press, New York.
- Boisvert D. P. and Schreiber C. (1992) Interrelationship of excitotoxic and free radical mechanisms, in Pharmacology of cerebral ischemia 1992 (Krieglstein J. and Oberpichler-Schwenk H., eds), pp. 311-320. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart.
- Buisson A., Plotkine M., and Boulu R. G. (1992a) The neuroprotective effect of a nitric oxide inhibitor in a rat model of focal cerebral ischaemia. Br. J. Pharmacol. 106, 766-767.
- Buisson A., Margaill I., Allix M., Callebert J., Plotkine M., and Boulu R. G. (1992b) Role of nitric oxide in focal cerebral ischemia, in Pharmacology of cerebral ischemia 1992 (Krieglstein J. and Oberpichler-Schwenk H., eds), pp. 417-425. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart.
- Choi D. W. (1992) Excitotoxic cell death. J. Neurobiol. 23, 1261-1276.
- Coyle J. T. and Puttfarcken P. (1993) Oxidative stress, glutamate and neurodegenerative disorders. Science 262, 689-695.
- Dawson, V. L., Dawson T. M., London E. D., Bredt D. S., and Snyder S. H. (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA 88, 6368-6371.
- Ginsberg M. D., Globus M. Y.-T., Martinez E., Morimoto T., Lin B., Schnippering H., Alonso O. F., and Busto R. (1994) Oxygen radical and excitotoxic processes in brain ischemia and trauma, in Pharmacology of cerebral ischemia 1994 (Krieglstein J. and Oberpichler-Schwenk H., eds), pp. 255-268. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart.
- Hall E. D., Andrus P. K., and Yonker P. A. (1993) Brain hydroxyl radical generation in acute experimental head injury. J. Neurochem. 60, 588-594.
- Halliwell B., Harparkash K., and Ingelman-Sundberg M. (1991) Hydroxylation of salicylate as an assay for hydroxyl radicals: a cautionary note. Free Rad. Biol. Med. 10, 439-441.
- Hammer B., Parker Jr. W. D., and Bennett Jr. J. P. (1993) NMDA receptors increase OH radicals in vivo by using nitric oxide synthase and protein kinase C. NeuroReport 5, 72-74.
- Iadecola C., Pelligrino D. A., Moskowitz M. A., and Lassen N. A. (1994) Nitric oxide synthase inhibition and cerebrovascular regulation. J. Cereb. Blood Flow Metab. 14, 175-192.
- Lafon-Cazal M., Pietri S., Culcasi M., and Bockaert J. (1993) NMDA-dependent superoxide production and neurotoxicity. Nature 364, 535-537.
- Lancelot E., Callebert J., Plotkine M., and Boulu R. G. (1997) Alpha-phenyl-N-tert-butylnitrone attenuates excitotoxicity in rat striatum by preventing hydroxyl radical accumulation. Free. Radic. Biol. Med 23, 1031-1034.
- Lancelot E., Revaud M.L., Boulu R. G., Plotkine M. and Callebert J. (1995) Striatal dopamine participates in glutamate-induced hydroxyl radical generation. NeuroReport 6, 1033-1036.
- Miyamoto M. and Coyle J. T. (1990) Idebenone attenuates neuronal degeneration induced by intrastriatal injection of excitotoxins. Exp. Neurol. 108, 38-45.
- Obata T. and Chiueh C. C. (1992) In vivo trapping of hydroxyl free radicals in the striatum utilizing intracranial microdialysis perfusion of salicylate: effects of MPTP, MPDP+, and MPP+. J. Neural Transm. (Gen. Sect.) 89, 139-145.
- Paxinos G. and Watson C. (1982) The rat brain in stereotaxic coordinates. Academic Press, Sydney.
- Phillis J. W. and Sen S. (1993) Oxypurinol attenuates hydroxyl radical production during ischemia/reperfusion injury of the rat cerebral cortex: an ESR study. Brain Res. 628, 309-312.
- Robinson T. E. and Whishaw I. Q. (1988) Normalisation of extracellular dopamine in striatum following recovery from partial unilateral 6-OHDA lesion of the substantia nigra: a microdialysis study in freely moving rats. Brain Res. 450, 209-224.
- Sun A. Y., Cheng Y., Bu Q., and Oldfield F. (1992) The biochemical mechanisms of the excitotoxicity of kainic acid. Mol. Chem. Neuropathol. 17, 51-62.
| Discussion Board | Previous Page | Your Poster Session |