Invited Symposium: Cerebral Artery Pharmacology and Physiology
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
During the last two decades it has become widely accepted that neuronal damage in acute neurodegenerative disease is due to a perturbation of cellular calcium metabolism (for recent reviews see Choi 1995, Kristián and Siesjö, 1996; Kristián and Siesjö, 1998). Insults, like ischemia, which compromise cell bioenergetic status, lead to cell depolarisation and to a rise in intracellular calcium concentration (Ca2+i). When ischemia is terminated by reperfusion the bioenergetic potential usually recovers and the ion gradients are re-established. However, in spite of that neuronal damage is observed following hours or days of recirculation (Kirino, 1982, Pulsinelli et al., 1982).
A marked and prolonged increase in Ca2+i is harmful to cells because it leads to activation of calcium-dependent enzymes having a potentially adverse effects, such as lipases, proteases, endonucleases and phosphatases. However, cell calcium overload can also cause mitochondrial failure, if this becomes irreversible it leads to cell death. Recently, the mitochondria have became the main focus of interest in cell death pathways known as apoptosis (for reviews see Kroemer et al., 1998; Green and Reed, 1998) and mitochondria probably also play a key role in delayed postischemic cell death (Siesjo et al., 1998a; 1998b). This is because during recovery re-energized mitochondria take up most of the calcium which has entered the cell during the insult. The uptake of calcium activates mitochondrial phospholipases, and seems to trigger increased production of free radicals (Dugan et al., 1995; Reynolds and Hastings, 1995; Stout et al., 1998) as well as release of mitochondrial proteins, some of which are proapoptogenic (Ouyang et al., 1998). The proapoptotic factors seem to be released when the mitochondrial membrane permeability is increased by the opening of a large conductance channel, the mitochondrial permeability transition (MPT) pore (Zamzami et al., 1996; Scarlett and Murphy, 1997). Thus, gradually raising Ca2+i followed by mitochondrial calcium accumulation and increased free radical production finally leads to irreversible damage to mitochondria, and to the triggering of a cell death program. In this review we will focus on some aspects of ischemia-induced and calcium-dependent mitochondrial dysfunction leading to the assembling of a mitochondrial transition pore.
1. Cell calcium metabolism and ischemia
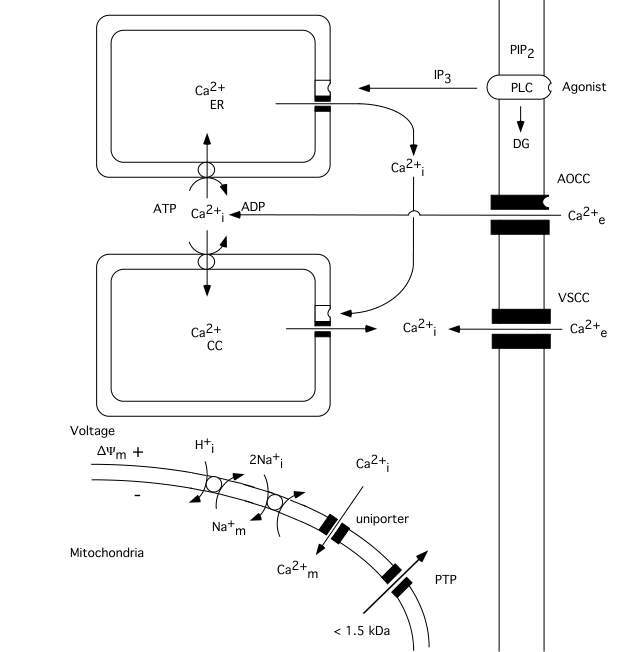
The calcium ion plays a key role as a regulator of numerous cellular functions. Therefore, cells tightly control the free intracellular calcium concentration (Ca2+i)(Carafoli, 1987; Miller, 1991; Berridge, 1997, 1998). Since the extracellular calcium concentration (Ca2+e) is several-fold higher (1mM) than the intracellular one (about 100 nM) even a small increase in the permeability of cell membranes to calcium ions lead to significant rise in Ca2+i. As fig. 1 shows calcium can enter cells via voltage- and agonist–operated calcium channels and can be released from intracellular stores, i.e. the endoplasmic reticulum (ER) or so called calciosomes. Calcium stores of ER origin are depleted either when inositol trisphophate (IP3) is formed by activated phospholipase C (PLC) or when a rise in Ca2+i triggers a release of calcium from IP3 non-sensitive stores via calcium induced calcium release (CICR) mechanisms. The mitochondria also represent a potential calcium source; however, during normal physiological conditions mitochondrial calcium content is low (abour 200 nM) in resting cells (Rizzuto et al. 1994; Babcock et al. 1997).
Mitochondria modulate the free cytosolic calcium concentration during and following intense activation of calcium conductances in plasma membranes (Friel and Tsien, 1994; White and Reynolds, 1996; White and Reynolds, 1997; Wang and Thayer, 1996). At steady state, there is a balance between influx and efflux of Ca2+ across the mitochondrial membrane. Mitochondria start to accumulate calcium when the cytosolic calcium concentration rises over a "set point" (about 500 nM). Ca2+ uptake by the mitochondria occurs via an uniporter (channel), and is driven by the mitochondrial membrane potential (*Ym). The rate of uptake is proportional to Ca2+i (for review see Gunter and Pfeiffer, 1990).
The efflux of Ca2+ from mitochondria can take place by to different mechanisms. After moderate calcium accumulation the Ca2+ ions are transported out from mitochondria via Ca2+/2Na+ exchange, with coupled Na+/H+ exchange to normalizing the Na+ gradient. However, when mitochondria are exposed to a sustained elevation of extramitochondrial calcium concentration a mitochondrial permeability transition (MPT) pore is apt to be opened. The MPT pore is a high conductance one since it is open to molecules with a molecular size up to 1500 Daltons. As a result the accumulated calcium is released, the mitochondria depolarize, and a large amplitude swelling of mitochondrial matrix occurs (Gunter and Pfeiffer, 1990; Zoratti and Szabó, 1995; Bernardi and Petronilli, 1996).
 Click to enlarge
Click to enlarge
Fig 1 Schematic diagram illustrating calcium fluxes associated with the plasma membrane, and endoplasmic reticulum, as well as with mitochondria. Influx across the plasma membrane occurs through voltage- and agonist operated calcium channels. Agonists acting on surface receptors coupled to phospholipase C (PLC) trigger the production of inositol trisphosphate (IP3) and diacylglyceride (DG). IP3 activates receptors on ER, triggering Ca2+ release. A rise in Ca2+i can then activate a calcium-induced calcium release from calciosomes. The lower part of figure shows the calcium fluxes across mitochondrial membranes. Mitochondria take up calcium via an electrogenic uniporter, while efflux occurs by Ca2+/Na+ exchange, with H+/Na+ exchange restoring the Na+ gradient. When the mitochondrial permeability transition (MPT) pore is opened solutes of molecular weight up to 1.5 kDa can be released from mitochondrial matrix. Slightly modified after Siesjö et al. 1999..
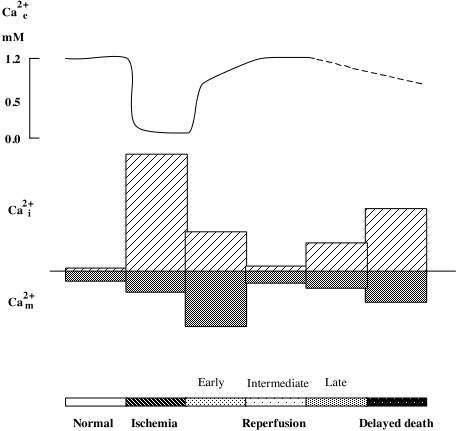
As mentioned above ischemia is accompanied by bioenergetical failure causing massive cell depolarisation, with efflux of K+ and uptake of Na+, Cl-, and Ca2+ (for review see Kristián and Siesjö, 1997). Fig 2 shows that the depolarisation causing downhill ion fluxes occurs after a short delay (about 1 min) following the onset of ischemia. During this period cells are using the available energy stores for fueling the membrane pumps. At the time of ischemic depolarisation extracellular calcium concentration (Ca2+e) is reduced from a baseline level of about 1.2 mM to about 0.1 mM.
Changes in Ca2+i during ischemia mirror the alterations in Ca2+e (Silver and Erecinska, 1990; Silver and Erecinska, 1992). During the first min of ischemia there is a small rise in Ca2+i (from about 70 nM to 150 nM) which is then followed by a marked increase to about 30 mMif ischemia is followed by optimal recirculation the phosphorylation potential recovers after about two min, and ion gradients are restored. Normalisation of Ca2+e takes place in two phases. First, a partial rapid recovery of Ca2+e occurs during the cell membrane repolarisation (up to about 70% of control values). This is followed by a slow, gradual increase towards the preischemic baseline level. The later probably reflects the extrusion of calcium which is secondarily released from ER or mitochondria.
Even if cells seem to survive the ischemic insult, cell death occurs after hours or days. In this reperfusion period there are sings of a perturbed cell calcium metabolism. For example, the total cell calcium content increases (Dienel, 1984; Deshpande et al., 1987; Young et al., 1986; Kristián et al., 1998). Also recording of Ca2+i during the recovery period revealed a delayed secondary rise in Ca2+i (Silver and Erecinska 1992). The intracellularly accumulated calcium progressive uptake of Ca2+ by the mitochondria (Dux et al., 1987; Zaidan and Sims, 1994). A high intramitochondrial calcium increases the probability of an MPT pore opening (see above). The immunosuppressant cyclosporin A (CsA) which efficiently blocks the pore, protects against the neuronal damage which is associated with forebrain ischemic insult (Uchino et al., 1995, 1998), suggesting that CsA blocks Ca2+ mediated pore opening.
 Click to enlarge
Fig 2 Schematic illustration of changes in extra-, intracellular Ca2+ concentration. (Ca2+e, Ca2+i ), and mitochondrial Ca2+ content (Ca2+m ) during ischemia and reperfusion period..
Click to enlarge
Fig 2 Schematic illustration of changes in extra-, intracellular Ca2+ concentration. (Ca2+e, Ca2+i ), and mitochondrial Ca2+ content (Ca2+m ) during ischemia and reperfusion period..
2. Mitochondria and ischemic cell death
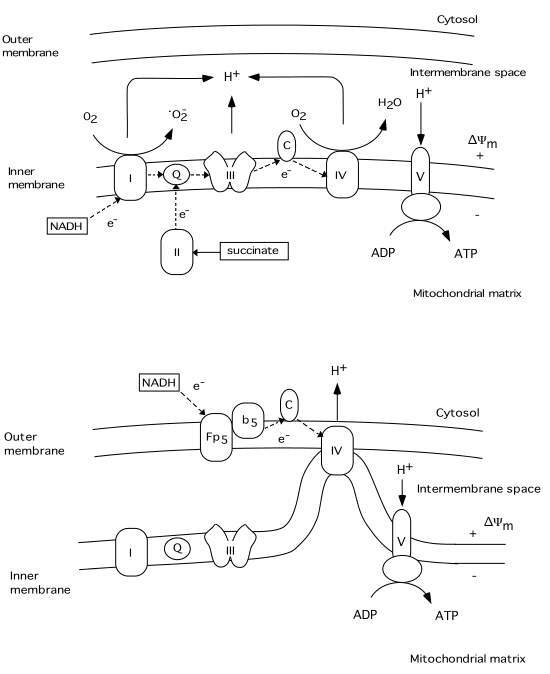
Under normal conditions electron transport in the mitochondrial respiratory chain creates both a H+ gradient across the inner mitochondrial membrane and an electrical potential, the inside of the mitochondria being negative. During this process reactive oxygen species (ROS) are produced (see fig.3). Up to 5 % of the oxygen reduced is converted by complex I to superoxide (oO2-)(Cadenas, 1989). It is known that the MPT pore can be induced by ROS probably due to dithiol cross-linking (Kowaltowski , 1998; Zamzami, 1998). Thus, increased production of superoxide favors the activation of a mitochondrial MPT pore. It appears that this pore can act at least in two different levels of conductance and reversibility. At low level of conductance the MPT pore opening is reversible and does not entail a large amplitude swelling of mitochondrial matrix, although it does cause a collapse of the *Ym (Ichas and Mazat, 1998). At high level of conductance, the MPT pore opening is irreversible and leads to large amplitude swelling of the mitochondrial matrix. During this event cytochrome c and the apoptosis inducing factor (AIF) are released from mitochondria (Kantrow and Piantadosi, 1997; Susin et al., 1997; Zamzami et al., 1997; Kluck et al., 1997). However, it is not clear if increased production of ROS precedes the MPT pore opening, or if the rise in ROS production is the result of the permeability change of the mitochondrial membranes.
For example, superoxide is produced by mitochondria due to a switch from the normal four-electron reduction of O2 to a one-electron reduction even when cytochrome c is released from mitochondria (Cai and Jones, 1998; see also Skulachev, 1998). As shown in fig 3 cytochrome c participates in electron transport from complex III (bc1 complex) to complex IV (cytochrome oxidase). However, it can also act as an antioxidant (see Skulachev, 1998). In this case the superoxide is converted back to oxygen and cytochrome c is reduced. The reduced from of cytochrome c is then oxidized either by complex IV or cytochrome c peroxidase localized in the intermembrane space (Gilmour et al., 1994). Following the release of cytochrome c the electron transport by respiratory complexes localized in the inner membrane is inhibited however, the respiration and electron transport can continue via respiratory enzymes located at external membrane sites(Skulachev, 1998; La Piana et al., 1998; see fig 3).
Thus, cytochrome c located on the external side of the outer membrane can activate an electron transport chain which promotes the transfer of electrons from substrate present in the cytosol to cytochrome oxidase inside the mitochondria. Therefore the mitochondrial membrane potential can be upheld and ATP can still be formed even though part of the pool of cytochrome c has been released. However, if the MPT pore is ireversibly opened the respiration is inhibited due to loss of pyridine nucleotides and substrates from mitochondria, blocking in turn also the citric acid cycle metabolism.
...as can be seen in Figure 3.
 Click to enlarge
Fig 3 Schematic illustration of the respiratory chain complexes under normal physiological conditions (upper panel) and after cytochrome c release into the cytosol (lower panel). A H+ gradient generated by complex I (NADH dehydrogenase), III (ubiquinol-cytochrome c reductase), and IV (cytochrome c oxidase) is used by complex V (ATP synthase) to generate ATP from ADP. During this process a superoxide (oO2-) is produced by complex I (Q = ubiquinone, C = cytochrome c). The lower panel illustrates possible mechanisms of continued respiration after cytochrome c is released from the intermembrane space to the cytosol. Tentatively, cytochrome c localized on the outer membrane can receive electrons from an external respiratory chain (Fp5 NADH cytochrome b5 reductase), bypassing complex I and III. The H+ gradient would then be generated by complex IV.
Click to enlarge
Fig 3 Schematic illustration of the respiratory chain complexes under normal physiological conditions (upper panel) and after cytochrome c release into the cytosol (lower panel). A H+ gradient generated by complex I (NADH dehydrogenase), III (ubiquinol-cytochrome c reductase), and IV (cytochrome c oxidase) is used by complex V (ATP synthase) to generate ATP from ADP. During this process a superoxide (oO2-) is produced by complex I (Q = ubiquinone, C = cytochrome c). The lower panel illustrates possible mechanisms of continued respiration after cytochrome c is released from the intermembrane space to the cytosol. Tentatively, cytochrome c localized on the outer membrane can receive electrons from an external respiratory chain (Fp5 NADH cytochrome b5 reductase), bypassing complex I and III. The H+ gradient would then be generated by complex IV.
Released cytochrome c and AIF activate the proapoptotic proteases-caspases whose activation leads to apoptotic cell death. However, activated caspases can also cause damage to mitochondrial membranes accompanied by irreversible swelling of mitochondria (Marzo et al., 1998). Furthermore, changes in energy metabolism can shift the balance between apoptosis and necrosis (Richter et al., 1996; Hirsch et al., 1997).
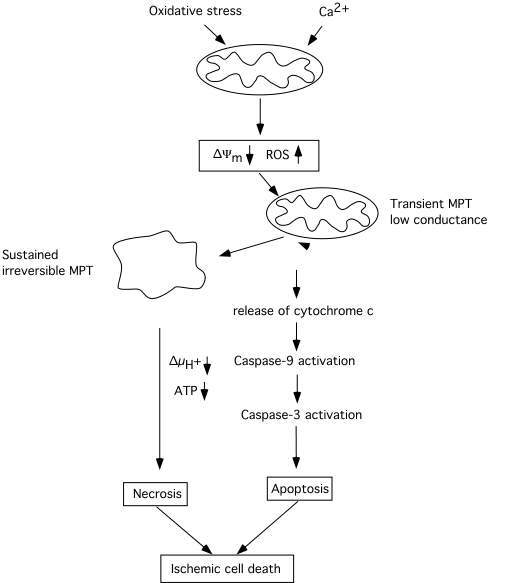
Fig 4 attempts to summary events which are triggered by mitochondrial dysfunction, involving a decrease in *Ym, probably also an enhanced production of ROS. The decrease in *Ym can trigger a transient MPT of low coductance which causes release of cytochrome c, activation of caspase 3, and apoptotic cell death. Caspase-3 activation may feed back to the mitochondria and cause further depolarisation and dysfunction, If the bioenergetic state is compromised, or massive amount of cytochrome c is released, the electrochemical potential for H+ is collapsed and ATP production ceases. The result is necrotic cell death. However, as pointed out by MacMannus and Linnik (1997) ischemic cell death may be an entity per se since it does not exclusively conform to either necrosis or apoptosis.
...as can be seen in Figure 4.
 Click to enlarge
Fig 4 Schematic diagram illustrating the cascade of events triggered by mitochondria which can lead to apoptotic/necrotic cell death following ischemic insult. A decrease of mitochondrial membrane potential due to calcium accumulation rises the production of ROS and triggers activation of an MPT and the release of cytochrome c. Activated caspases can act as positive feedback leading to further damage to mitochondrial membranes and to an irreversible MPT pore opening accompanied by mitochondrial matrix swelling. Extensive bionergetical failure due to dysfunction of a large part of the mitochondrial population within the cell can activate necrotic processes which do not require ATP. Modified after Siesjö et al. (1999).
Click to enlarge
Fig 4 Schematic diagram illustrating the cascade of events triggered by mitochondria which can lead to apoptotic/necrotic cell death following ischemic insult. A decrease of mitochondrial membrane potential due to calcium accumulation rises the production of ROS and triggers activation of an MPT and the release of cytochrome c. Activated caspases can act as positive feedback leading to further damage to mitochondrial membranes and to an irreversible MPT pore opening accompanied by mitochondrial matrix swelling. Extensive bionergetical failure due to dysfunction of a large part of the mitochondrial population within the cell can activate necrotic processes which do not require ATP. Modified after Siesjö et al. (1999).
References
- Babcock, D. F., Herrington, J., Goodwin, P. C., Park, Y. B., and Hille, B. (1997). Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol 136, 833-44.
- Bernardi, P., and Petronilli, V. (1996). The permeability transition pore as a mitochondrial calcium release channel: a critical apppraisal. J Bioener and Biomem 28, 131-137.
- Berridge, M. J. (1998). Neuronal calcium signalling. Neuron 21, 13-26.
- Berridge, M. J. (1993). A tale of two messengers. Nature 365, 388-391.
- Cadenas, E. (1989). Biochemistry of oxygen toxicity. Ann. Rev. Biochem. 58, 79-110.
- Cai, J., and Jones, D. P. (1998). Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 273, 11401-4.
- Carafoli, E. (1987). Intracellular calcium homeostasis. Ann Rev Biochem 56, 395-433.
- Choi, D. (1995). Calcium: still center-stage in hypoxic-ischemic neuronal death. TINS 18, 58-60.
- Deshpande, J. K., Siesjö, B. K., and Wieloch, T. (1987). Calcium accumulation and neuronal damage in the rat hippocampus following cerebral ischemia. J. Cereb. Blood Flow Metab. 7, 89-95.
- Dienel, G. (1984). Regional accumulation of calcium in postischemic rat brain. J Neurochem 43, 913-925.
- Dugan, L., Sensi, S., Canzoniero, L., Handran, S., Rothman, S., Lin, T.-S., Goldberg, M., and Choi, D. (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neuroscience 15, 6377-6388.
- Dux, E., Mies, G., Hossmann, K.-A., and Siklos, L. (1987). Calcium in the mitochondria following brief ischemia of gerbil brain. Neurosci Lett 78, 295-300.
- Friel, D. D., and Tsien, R. W. (1994). An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci 14, 4007-24.
- Gilmour, R., Goodhew, C. F., Pettigrew, G. W., Prazeres, S., Moura, J. J., and Moura, I. (1994). The kinetics of the oxidation of cytochrome c by Paracoccus cytochrome c peroxidase. Biochem J 300, 907-14.
- Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309-12.
- Gunter, T., and Pfeiffer, D. (1990). Mechanisms by which mitochondria transport calcium. Am J Physiol 258, C755-786.
- Hirsch, T., Marchetti, P., Susin, S. A., Dallaporta, B., Zamzami, N., Marzo, I., Geuskens, M., and Kroemer, G. (1997). The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 15, 1573-81.
- Ichas, F., and Mazat, J. P. (1998). From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta 1366, 33-50.
- Kantrow, S. P., and Piantadosi, C. A. (1997). Release of cytochrome c from liver mitochondria during permeability transition. Biochem Biophys Res Commun 232, 669-71.
- Kirino, T. (1982). Delayed neuronal death in the gerbil hippocampus following transient ischemia. Brain Res 239, 57-69.
- Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer, D. D. (1997). The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis [see comments]. Science 275, 1132-6.
- Kristián, T., Gido, G., Kuroda, S., Schutz, A., and Siesjo, B. K. (1998). Calcium metabolism of focal and penumbral tissues in rats subjected to transient middle cerebral artery occlusion. Exp Brain Res 120, 503-9.
- Kristián, T., and Siesjö, B. (1996). Calcium-related damage in ischemia. Life Sci 59, 357-367.
- Kristián, T., and Siesjö, B. (1997). Changes in ionic fluxes during cerebral ischemia. In Neuroprotective agents and cerebral ischemia, A. Cross and A. Green, eds. (New York: Academic Press), pp. 27-45.
- Kristián, T., and Siesjo, B. K. (1998). Calcium in ischemic cell death. Stroke 29, 705-18.
- Kroemer, G., Dallaporta, B., and Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60, 619-42.
- La Piana, G., Fransvea, E., Marzulli, D., and Lofrumento, N. E. (1998). Mitochondrial membrane potential supported by exogenous cytochrome c oxidation mimics the early stages of apoptosis. Biochem Biophys Res Commun 246, 556-61.
- Li, Y., Chopp, M., Jiang, N., Zhang, Z., and Zaloga, C. (1995). Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke 26, 1252-1258.
- MacManus, J. P., and Linnik, M. D. (1997). Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab 17, 815-32.
- Marzo, I., Susin, S. A., Petit, P. X., Ravagnan, L., Brenner, C., Larochette, N., Zamzami, N., and Kroemer, G. (1998). Caspases disrupt mitochondrial membrane barrier function. FEBS Lett 427, 198-202.
- Miller, R. J. (1991). The control of neuronal Ca2+ homeostasis. Prog. Neurobiol 37, 255-285.
- Ouyang, Y. B., Siesjo, B. K., and Zivin, J. A. (1998). Release of cytochrome c from mitochondria in neurons following transient cerebral ischemia in rats. In Neuroscience Meeting (Los Angeles, pp. 1842.
- Pulsinelli, W. A., Brierley, J. B., and Plum, F. (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 11, 491-498.
- Reynolds, I., and Hastings, T. (1995). Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci 15, 3318-3327.
- Richter, C., Schweizer, M., Cossarizza, A., and Franceschi, C. (1996). Control of apoptosis by the cellular ATP level. FEBS Lett 378, 107-10.
- Rizzuto, R., Bastianutto, C., Brini, M., Murgia, M., and Pozzan, T. (1994). Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol 126, 1183-94.
- Scarlett, L. J., and Murphy, M. P. (1997). Release of apoptogenic proteins from the mitochondrial intermembrane space during the mitochondrial permeability transition. FEBS Letters 418, 282-286.
- Siesjo, B. K., Eskil, E., Janelidze, S., Keep, M.,Kristian, T., Ouyang, Y., and Uchino, H.(1998). Role and mechanisms of secondary mitochondrial failure. Acta Neuroscit in press.
- Siesjö B.K., Hu B.R., and Kristián ., (1999) Is the cell death triggered by the mitochondria or the endoplasmic reticulum. Journal of cerebral blood flow and Metab, in press
- Silver, I., and Erecinska, M. (1990). Intracellular and extracellular changes of [Ca2+] in hypoxia and ischemia in rat brain in vivo. J Gen Physiol 95, 837-866.
- Silver, I. A., and Erecinska, M. (1992). Ion homeostasis in rat brain in vivo: intra- and extracellular Ca2+ and H+ in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab 12, 759-772.
- Skulachev, V. P. (1998). Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett 423, 275-80.
- Stout, A. K., Raphael, H. M., Kanterewicz, B. I., Klann, E., and I.J., Reynolds (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nature Neuroscience 1, 366-373.
- Susin, S. A., Zamzami, N., Larochette, N., Dallaporta, B., Marzo, I., Brenner, C., Hirsch, T., Petit, P. X., Geuskens, M., and Kroemer, G. (1997). A cytofluorometric assay of nuclear apoptosis induced in a cell-free system: application to ceramide-induced apoptosis. Exp Cell Res 236, 397-403.
- Uchino, H., Eskil, E., Uchino, K., Lindvall, O., and Siesjö, B. (1995). Cyclosporin A dramatically ameliorates CA1 hippocampal damage following transient forebrain ischemia in the rat. Acta Physiol Scand 155, 469-471.
- Uchino, H., Eskil, E., Uchino, E., Li, P.A., He, Q.P., Smith, M.L., Siesjö, B.K. (1998) Amelioration by cyclosporine A of brain damage in transient forebrain ischemia in the rat. Brain Res, in press.
- Wang, G. J., and Thayer, S. A. (1996). Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol 76, 1611-21.
- White, R. J., and Reynolds, I. J. (1997). Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol 498, 31-47.
- White, R. J., and Reynolds, I. J. (1996). Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci 16, 5688-97.
- Young, W., DeCrescito, V., Flamm, E. S., Hadani, M., Rappaport, H., and Cornu, P. (1986). Tissue Na, K, and Ca changes in regional cerebral ischemia: their measurement and interpretation. Cent Nerv Syst Trauma 3, 215-34.
- Zaidan, E., and Sims, N. (1994). The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. J Neurochem 63, 1812-1819.
- Zamzami, N., Hirsch, T., Dallaporta, B., Petit, P. X., and Kroemer, G. (1997). Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr 29, 185-93.
- Zamzami, N., Susin, S. A., Marchetti, P., Hirsch, T., Gomez-Monterrey, I., Castedo, M., and Kroemer, G. (1996). Mitochondrial control of nuclear apoptosis. J Exp Med 183, 1533-1544.
- Zoratti, M., and Szabó, I. (1995). The mitochondrial permeability transition. Biochimica et Biophysica Acta 1241, 139-176.
| Discussion Board | Previous Page | Your Symposium |