Invited Symposium: Medicinal Plants and Drug Actions
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
INTRODUCTION
Tetrandrine is an alkaloid isolated from the Chinese medical herb Radix stephania tetrandrae. It has been used clinically in China for many years to treat angina and hypertension with good results (1). Recent pharmacological studies have demonstrated that tetrandrine blocks inward calcium current through the voltage-sensitive L-type calcium channel (2). Since it competitively inhibits [3H]-diltiazem binding, partly inhibits [3H]- verapamil binding and stimulates [3H]-nitrendipine binding to cardiac sarcolemmal membranes in a manner very similar to diltiazem, it is likely that tetrandrine acts at the benzothiazepine site of the L-type calcium channel (3).
Chemically, tetrandrine is a bis-benzyl-tetrahydroisoquinoline with its 2 subunits connected in a head-to-head, tail-to-tail fashion through ether linkages (4). Chemical synthesis of tetrandrine is difficult because of its complicated structure. The pharmacological profile of tetrandrine indicates that in comparison to the dihydropyridine calcium channel blockers, tetrandrine has less potency and selectivity for blood vessels (5). Therefore, the clinical use of tetrandrine has been somewhat limited. In order to simplify the chemical structure of tetrandrine and find better therapeutic agents, we have synthesized and screened, using both [3H]-nitrendipine binding and biological assays, a series of benzyl-tetrahydroisoquinolines (half-molecules of tetrandrine). CPU-23 (1-{1-[(6-methoxy)-naphth-2-yl]}-ethyl-2-(1-piperidinyl)-acetyl-6,7-dimethoxy- 1,2,3,4-tetrahydroisoquinoline), the most potent compound identified in this series (4), was found to inhibit KCl-induced contraction of rat aorta and displace [3H]-nitrendipine binding in rat cerebral cortical membranes with similar potency. It was also found that CPU-23 behaves as a simple competitive inhibitor at the [3H]-nitrendipine binding sites (6). Whole-cell patch clamp experiments revealed that CPU-23 dose-dependently blocked L-type Ca2+ channels and delayed rectifier K+ channels in single smooth muscle cells isolated from rat tail artery and rabbit portal vein. In addition, it inhibited KCl-induced Ca2+ influx and action potential characteristics of myocardial preparation in vitro and induced hypotension and bradycardia in rat in vivo. It is indicated that CPU- 23 is a novel potent Ca2+ and delayed rectifier K+ channels blocker, which has therapeutic potentials of antihypertension and antiarrhythmics (6,7). This review summarizes in detail the cardiovascular pharmacology of CPU-23.
IN VITRO PHARMACOLOGY
[3H]-Nitrendipine Binding Studies
With the availability of radioactive ligands of dihydropyridines, verapamil and diltiazem at high specific activity, it has become possible to study drug interactions at the L-type calcium channel by direct radioligand binding assays. Such studies indicate that these compounds interact at distinct binding sites which are allosterically linked on the channel complex and that occupation of one site affects ligand binding at other sites. For instance, the specific binding of [3H]- nitrendipine to cardiac sacrolemma and brain membrane was competitively inhibited by nifedipine, only partly inhibited by verapamil and was allosterically stimulated by diltiazem and tetrandrine (3,8). Unexpectedly, in our [3H]-nitrendipine binding study, unlike tetrandrine which stimulated [3H]-nitrendipine binding, none of the substituted tetrahydroisoquinolines exhibited a stimulatory effect. Instead, many of them inhibited [3H]-nitrendipine binding to rat cerebral cortical membranes in a concentration-dependent manner (Table 1). From the relative potencies of substituted tetrahydroisoquinalines in inhibiting [3H]-nitrendipine binding, several structure-activity features can be deduced (for details see ref. 6).
Table 1. Inhibition of specific [3H]-nitrendipine binding to rat cerebral cortical membranes and 80 mM KCl-induced contraction of rat aortic strips by classical calcium entry blockers and tetrahydroisoquinolines
________________________________________________________________________
IC50 (M)
_________________________________________________________
Drugs Binding Relaxation of aorta
________________________________________________________________________
Nifedipine 1.1�0.2�10-9 1.2�0.3�10-9
Verapamil 2.1�0.1�10-7 6.3�0.5�10-8
Diltiazem Stimulation* 1.4�0.2�10- 7
Tetrandrine Stimulation* 7.0�1.9�10-7
CPU-21 4.6�0.6�10-6 2.5�0.3�10-6
CPU-23 5.1�0.8�10-7 2.1�0.2�10-7
CPU-50 7.6�1.0�10-7 5.0�0.4�10-7
CPU-57 4.1�0.1�10-7 2.5�0.3�10-7
________________________________________________________________________
IC50 is the molar concentration of drug required to give 50% inhibition of specific [3H]-nitrendipine binding or 80 mM KCl-induced contraction of rat aortic strips. Results are the mean � s.e.mean of 3 (for binding) and 6 (for bioassay) separate experiments. *Diltiazem (10-5 M) and tetrandrine (10-5 M) enhanced specific [3H]-nitrendipine binding at 37oC to 168 � 8% and 263 � 29% of control, respectively. The same concentration of diltiazem did not stimulate [3H]-nitrendipine binding at 22° (103 � 3%) while tetrandrine was still able to enhance binding to 154 � 6% of control.
CPU-23 was one of the most potent of the tetrahydroisoquinoline analogues identified in this series with an IC50 value of 0.51 �M. When the displacement curve was analyzed with the Hill plot, the Hill coefficient was found to close to unity, suggesting that CPU-23 may act as simple competitive inhibitor at the nitrendipine binding site of the L-type calcium channel. To evaluate further the nature of the interaction with the dihydropyridine site, we analyzed the effects of various concentrations of CPU-23 on the Kd (equilibrium dissociation constant) and Bmax (maximum number of binding sites) of [3H]-nitrendipine binding by saturation analysis. As illustrated in Table 2, CPU-23 behaved as a simple competitive inhibitor like nifedipine and significantly reduced the apparent affinity without affecting the Bmax of [3H]-nitrendipine binding. When the original data for CPU-23 were further analyzed by the Arunlakshana & Schild plot, a slope close to unity (0.94) and a KB value of 0.17 �M was obtained. This KBvalue agrees quite well with its Kivalue (0.26 �M) calculated from the drug displacement experiments by the Cheng-Prusoff equation (6). Further experiments to examine the effect of CPU-23 on the dissociation kinetics of [3H]- nitrendipine will provide a clear answer to the competitive nature of this interaction.
Table 2. Scatchard analysis of the interaction of CPU-23 with the binding of [3H]-nitrendipine to rat cerebral cortical membranes___________________________________________________________________ Conc. Kd Bmax Hill coefficient (�M) (nM) (fmol mg-1 protein) (nH) ________________________________________________________________________ 0 0.31�0.03 (6) 18.23�1.15 (6) 0.81�0.04 (6) 0.1 0.48�0.07 (4)* 16.49�1.45 (4) 0.83�0.04 (4) 0.3 1.15�0.09 (3)** 20.11�2.74 (3) 0.75�0.09 (3) 1 1.81�0.20 (3)** 15.47�1.79 (3) 1.00�0.05 (3) ________________________________________________________________________[3H]-nitrendipine (0.06-3 nM) was incubated with rat cerebral cortical membranes in the presence or absence of CPU-23 for 30 min at room temperature and specific binding was measured. Kd and Bmax were determined by Scatchard analysis with the LIGAND computer programme (Elsevier-BIOSOFT). Data shown are the mean � s.e.mean of (n) separate experiments performed in duplicate. *P < 0.05, **P < 0.001 when compared with control.
Effects on the Contractility of Blood Vessels
In a similar manner as the other prototypic calcium channel blockers, CPU-23 produced concentration-dependent inhibition of the contraction of rat aorta induced by 80 mM KCl with a pD2 value of 6.68. The rank order of potencies in this bioassay was: nifedipine > verapamil > diltiazem = CPU-23 > tetrandrine. Although CPU-23 was 175 times less potent than nifedipine, it was about 3 times more potent than tetrandrine in this test (Table 1). There is good correlation (r=0.99, p< 0.001) between the potency of substituted tetrahydroisoquinolines to inhibit specific [3H]-nitrendipine binding to rat cerebral cortical membranes and their ability to inhibit the KCl-induced contraction of rat aorta (Fig 1). This further supports the notion that substituted tetrahydroisoquinolines relax arterial smooth muscle by blocking the L-type calcium channels through interaction with the dihydropyridine site.
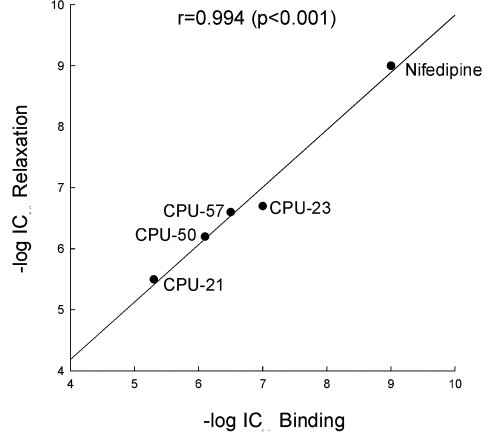 Fig. 1: Correlation between antagonism of 80 mM KCl-induced contraction of rat isolated aortic strips and inhibition of specific [3H]-nitrendipine binding by nifedipine and tetrahydro- isoquinolines. The correlation coefficient (r=0.99, P<0.001) was calculated by linear regression analysis.
Fig. 1: Correlation between antagonism of 80 mM KCl-induced contraction of rat isolated aortic strips and inhibition of specific [3H]-nitrendipine binding by nifedipine and tetrahydro- isoquinolines. The correlation coefficient (r=0.99, P<0.001) was calculated by linear regression analysis.
CPU-23 (1-10 �M) dose-dependently reduced concentration-response curves for KCl and phenylephrine (PE) in the rat tail artery. However, inhibition of KCl-induced contraction was much more potent than for PE: the maximal response to KCl was reduced to 51 � 5% and 28 � 4%, but the maximal response to PE was reduced to 85 � 6% and 78 � 5% by CPU-23 at the concentration of 1 and 10 �M (Fig 2). In isolated guinea-pig middle cerebral artery (in the presence of NG-nitro-L-arginine and indomethacin to prevent the production of NO and cyclo- oxygenase metabolites in endothelial cells), CPU-23 produced concentration-dependent relaxation of 80 mM KCl- or histamine-induced contraction with pD2 values of 6.60 and 7.04, respectively. When compared with CPU-23, nimodipine, a dihydropyridine with high selectivity for cerebral arteries, was more potent in the same tissue with pD2 values of 9.06 and 9.48 for relaxing 80 mM KCl- and histamine-induced contractions, respectively. The spontaneous contractions of the rat portal vein are dependent on extracellular calcium as they are abolished in calcium-free, or 2 mM EGTA containing physiological solution and are restored upon readministration of 2.5 mM Ca2+, or washout of EGTA. CPU-23 shared with verapamil and diltiazem the ability to inhibit the spontaneous contractions of the rat portal vein in a concentration manner (H. Dong, unpublished data). These data suggest that the vasorelaxation activity of CPU-23 is not limited to peripheral arteries and not dependent upon endothelial-derived mediators, but is effective in cerebral resistance vessels and results from the inhibition of extracellular calcium entry into the vascular smooth muscle cells.
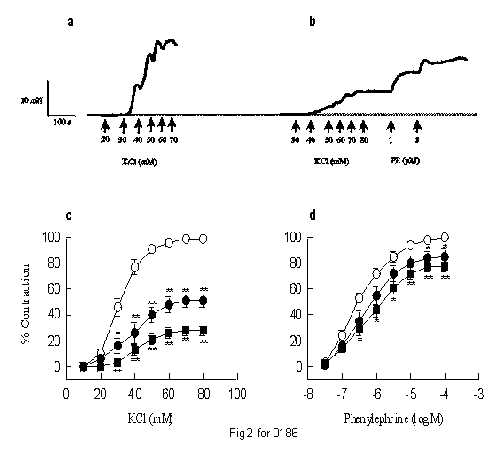 Fig. 2: Original tracings show the concentration-response curves of the rat tail artery for KCl and phenylephrine in the absence (a) and the presence of CPU-23 (10 �M, b). CPU-23 mediated inhibition of the concentration-response curves for KCl (c) and phenylephrine (d), plotted as mean � s.e. means from six to seven seperate experiments. *P < 0.05; **P < 0.01 compared to control.
Fig. 2: Original tracings show the concentration-response curves of the rat tail artery for KCl and phenylephrine in the absence (a) and the presence of CPU-23 (10 �M, b). CPU-23 mediated inhibition of the concentration-response curves for KCl (c) and phenylephrine (d), plotted as mean � s.e. means from six to seven seperate experiments. *P < 0.05; **P < 0.01 compared to control.
Actions on Ion Channels of Vascular Smooth Muscle
To determine if the inhibitory effects of CPU-23 observed on the blood vessels were due to direct effects of the drug on voltage-gated calcium channels (VGCCs), we undertook voltage clamp experiments using the perforated patch clamp technique. Utilizing freshly dispersed vascular smooth muscle cells from the rat tail artrey, whole cell barium currents were recorded in the absence and presence of CPU-23 (1 - 10 �M). The inward currents displayed characteristics typical of the voltage gated L-type calcium ion channel in that the currents were inhibited in the presence of the dihydropyridine calcium channel blocker nifedipine (0.1 �M) and potentiated with the dihydropyridine calcium channel activator Bay K 8644 (1 �M). We observed a dose-dependent decrease in the amplitude of the dihydropyridine-sensitive VGCC; CPU-23 (1 �M) reduced peak current by 30.9 � 6% wheres at 10 �M, peak current was reduced by 80.4 � 14% (Fig 3). Similar dose-dependent effects were also observed in isolated cells from the rabbit portal vein. These data provide direct evidence, indicating that CPU-23 can inhibit the opening of L-type VGCCs on vascular smooth muscle cells (9).
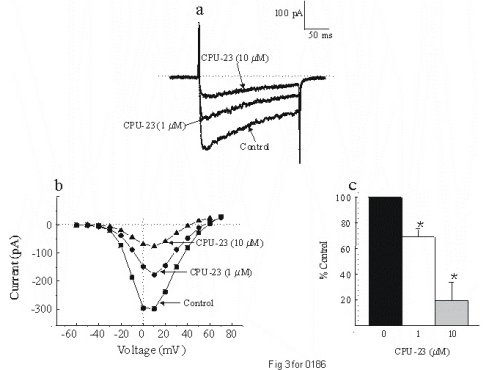 Fig. 3: Inward currents recorded using Ba2+ (10 mM), in freshly dispersed vascular smooth muscle cells from the rat tail artery. (a) Represensitive traces showing the effects of CPU-23 on the inward current elicited by stepping to 0 mV from a holding potential of -80 mV. A dose-dependent inhibition of the current was observed. (b). Representative tace showing the inhibitory effects of CPU-23 (1 and 10 �M) on the I-V relation for this current. (c) Bar graph showing inhibition of the inward current with CPU-23 plotted as mean � s.e. means from six to nine cells at +10 mV, the potential at which peak current was observed in all cells. *P < 0.05 compared to control.
Fig. 3: Inward currents recorded using Ba2+ (10 mM), in freshly dispersed vascular smooth muscle cells from the rat tail artery. (a) Represensitive traces showing the effects of CPU-23 on the inward current elicited by stepping to 0 mV from a holding potential of -80 mV. A dose-dependent inhibition of the current was observed. (b). Representative tace showing the inhibitory effects of CPU-23 (1 and 10 �M) on the I-V relation for this current. (c) Bar graph showing inhibition of the inward current with CPU-23 plotted as mean � s.e. means from six to nine cells at +10 mV, the potential at which peak current was observed in all cells. *P < 0.05 compared to control.
Three types of calcium channel blockers widely used clinically, namely the dihydropyridines (e.g. nifedipine, nitrendipine and nimodipine), the phenylalkylamines (e.g. verapamil) and benzothiazepines (e.g. diltiazem), have been reported to block K+ channels, which are membranes of the same supergene family as Ca2+ channels. We have also examined whether CPU- 23 acts on K+ channels like three types of calcium channel blockers and to explore its mechanism of action. Whole cell patch-clamp experiments revealed that CPU-23 dose- dependently decreased outward K+ currents with mainly blocking delayed rectifier K+ channels (Kdr) in rabbit vascular smooth muscle cells. Both end- pulse and tail delayed rectifier K+ currents (IdK) on depolarization from -80 to 30 mV and repolarization to -40 mV were reduced by CPU-23 with IC50 =3.5 � 0.3 �M at 10 mV, and with Hill coefficient of 0.47 � 0.02 (Fig 4). CPU-23 started to inhibit IdK at approximately 1 min and reached maximal inhibition at 4-5 min exposure, which was reversed at about 10 min washout. Tail currents recorded by depolarizing cells from -60 mV to 10 mV and then repolarizing to -40 mV, did not cross over each other in the absence and the presence of CPU-23. Taken together, these data support the idea that CPU-23 is also a potent reversible blocker of Kdr, which acts predominantly as a closed channel blocker of Kdr in rabbit vascular smooth muscle cells (Dong et al.,unpublished data).
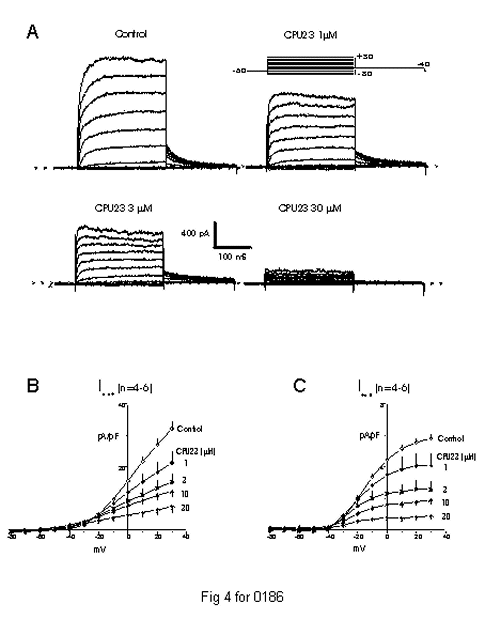 Fig. 4: Dose-dependent inhibition of delayed rectifier currents of rabbit portal vein myocytes by CPU-23. A: Representative families of whole cell currents recorded during 250-ms pulses to between -80 and +30 mV in 10-mV steps and tail currents during subsequent steps to -40 mV applied every 10 s from single myocytes before (control) and after exposure to CPU-23 (1-30 �M). B and C: Average current-voltage (I-V) relation for net (I net) and tail (I tail) currents in control conditions and after CPU-23 (1-30 �M) treatments in foure-six myocytes.
Fig. 4: Dose-dependent inhibition of delayed rectifier currents of rabbit portal vein myocytes by CPU-23. A: Representative families of whole cell currents recorded during 250-ms pulses to between -80 and +30 mV in 10-mV steps and tail currents during subsequent steps to -40 mV applied every 10 s from single myocytes before (control) and after exposure to CPU-23 (1-30 �M). B and C: Average current-voltage (I-V) relation for net (I net) and tail (I tail) currents in control conditions and after CPU-23 (1-30 �M) treatments in foure-six myocytes.
Cardiac Effects
In Langendorff perfused rat hearts, CPU-23, nifedipine and diltiazem decreased, in a dose- dependent manner, the heart rate with the potency of nifedipine being less compared with the other two drugs. Both nifedipine and diltiazem markedly inhibited the force of contraction in a concentration-dependent manner. On the other hand, the effect of CPU-23 was biphasic, inhibiting the developed tension at 0.1- 1 �M but enhancing the tension at 10 �M. Nifedipine possesses a more potent negative inotropic effect than negative chronotropic effect and causes cardiac arrest at concentrations higher than 0.3 �M. Interestingly, CPU-23 has a more potent negative chronotropic effect than negative inotropic effect and increased cardiac contraction at concentrations higher than 10 �M (7).
In electrophysiological studies, CPU-23 decreased action potential amplitude (APA) and the maximum upstroke velocity (Vmax ) and shortened the action potential duration at 50% and 90% of repolarization (APD50 and APD90) of action potentials in guinea-pig papillary muscles, though only at a concentration of 100 �M, and was without effect on resting potential (RP) and overshoot (OS). However, CPU-23 dose-dependently (1-100 �M) decreased APA, OS and Vmax and APD90 of slow action potentials induced by histamine in K+-depolarized guinea-pig papillary muscle (Table 3), where the action potential configuration is very dependent on calcium entry. In rabbit sinoatrial node tissue, where the action potential configuration is also dependent on calcium entry, CPU-23 decreased the APA, spontaneous rate and slope of phase 4 depolarization (dV/dt) in a dose-dependent manner (4).
Table3. Effects of CPU-23 on slow action potential characteristics in guinea-pig papillary muscles________________________________________________________________________
MDP (mV) APA (mV) OS (mV) APD90(ms) Vmax(Vs-1)
________________________________________________________________________
Control 56.2�5.4 94.6�4.1 33.0�3.1 177.0�13.2 11.0�0.5
CPU-23(�M)
1 54.8�5.7 84.4�2.9* 28.8�4.3* 168.6�11.8* 10.2�0.6*
10 53.0�3.0 73.0�2.6* 27.7�3.9* 145.0�3.0* 7.3�0.3**
100 50.7�3.1 67.0�6.5** 17.7�3.9** 84.3�4.3** 4.3�0.6**
________________________________________________________________________Data shown are the mean � s.e.mean of 5 separate experiments. MDP: maximum diastolic potential; APA: amplitude of action potential; OS: overshoot; APD90: action potetntial duration at 90% of repolarization; Vmax: maximum upstroke velocity. *P < 0.05; **P< 0.01 vs control.
KCl dose-dependently increases cytosolic free Ca2+ concentration [Ca2+ ]i in fura-2 loaded rat ventricular myocytes only in a medium containing calcium, and this effect is attenuated by nifedipine. By contrast, caffeine increases [Ca2+ ]i even in the absence of extracellular calcium. This suggests that KCl-induced increase in [Ca2+ ]i is due to extracellular calcium influx through L-type calcium channels whereas caffeine-induced increase in [Ca2+]i is dependent on intracellular Ca2+ mobilization. CPU-23 (1-10 �M) inhibited the KCl-induced increase in [Ca2+ ]i of rat ventricular myocytes significantly, but had no significant effect on the caffeine-induced increase in [Ca2+ ]i of rat ventricular myocytes in the calcium-free medium. These findings provide direct evidence that CPU-23 selectively inhibits calcium entry in ventricular myocytes (4,7).
IN VIVO PHARMACOL
Antihypertensive Effects after i.v. Injection
In pentobarbitone-anaesthetized Sprague-Dawley (SD) rats, acute administration of CPU-23 (1- 10 mg kg-1, i.v.) caused a rapid-onset and dose-dependent decrease in mean arterial blood pressure (MAP) and heart rate (HR). The hypotensive and bradycardic effects were rather slight and short-lived at 1 mg kg-1, but more pronounced and longer lasting at 3-10 mg kg-1, which remained significantly depressed even at 60 min after drug administration (Fig 5). Nifedipine, at a dose of 0.3 mg kg-1 i.v. produced a comparable decease in MAP to that of 5 mg kg-1 CPU-23, indicating CPU-23 is about 20 times less active than nifedipine in lowering blood pressure in SD rats. However, CPU-23 is about twice as active as tetrandrine in lowering blood pressure (10). Interestingly, when compared to nifedipine, CPU-23 is over 500 times less active in inhibiting [3H]-nitrendipine binding and is 175 times less potent in relaxing high K+-induced tension of rat aorta in vitro, but is only 20 times less active in lowering rat blood pressure in vivo. It is suggested that CPU-23 may have a better bioavailability than nifedipine in vivo or may induce hypotension via other modes of action. CPU-23 also differed from nifedipine in its effect on HR. At doses which caused a similar degree of hypotension, CPU-23 exerted a pronounced negative chronotropic action whereas nifedipine induced a reflex tachycardia. The marked bradycardia produced by CPU-23 may offer some myocardial protection in treating hypertensive conditions accompanying with a high circulatory concentration of catecholamines (11).
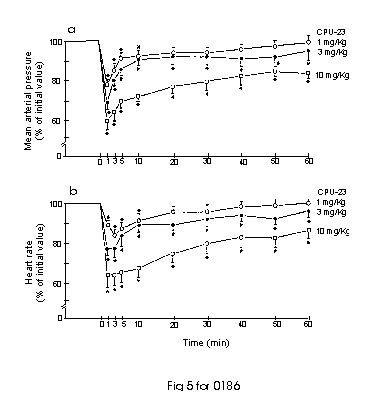 Fig. 5: Effects of CPU-23 i.v. on (a) mean arterial blood pressuure and (b) heart rate in pentobarbital-anaesthetized Sprague-Dawley rats. The drug (1 mg/k ( ), 3 mg/kg ( ) and 10 mg/kg ( )) was admistered at 0 min. Each point represents the mean of 5 animals; vertical bar show s.e. mean. *P< 0.05 when compared to control at zero time. Baseline: MAP=105 � 5 mmHg, HR=356� 12 beats/min.
Fig. 5: Effects of CPU-23 i.v. on (a) mean arterial blood pressuure and (b) heart rate in pentobarbital-anaesthetized Sprague-Dawley rats. The drug (1 mg/k ( ), 3 mg/kg ( ) and 10 mg/kg ( )) was admistered at 0 min. Each point represents the mean of 5 animals; vertical bar show s.e. mean. *P< 0.05 when compared to control at zero time. Baseline: MAP=105 � 5 mmHg, HR=356� 12 beats/min.
Similar to the observation in SD rats, intravenous bolus injection of CPU-23 produced a dose- dependent hypotensive and bradycardic response in both pentobarbitone-anaesthetized spontaneously hypertensive (SHR) and normotensive WKY rats. The extent and duration of the hypotension or bradycardia induced by CPU-23 were similar in SHR and WKY rats (6).
Antihypertensive Effects after i.c.v. InjectionAlthough the cardiovascular effects of calcium channel blockers are attributed mainly to a direct peripheral vasodilation and cardiac depression, recent findings suggest that they can exert indirect cardiovascular effects via interaction with the dihydropyridine receptor sites in the brain (12,13). CPU-23, at concentrations of 0.2-0.5 mg kg-1, which caused no significant cardiovascular effects after i.v. injection, induced a slow-onset, long-lasting and dose-dependent decrease of MAP and HR after intracerebroventricular (i.c.v.) injection, which was similar to the response to nifedipine at dose of 0.05 mg kg-1. After bilateral cervical vagotomy, the hypotensive effect of CPU-23 (0.5 mg kg-1) was significantly attenuated (14), suggesting that these effects of CPU-23 are most likely of central origin, as were those of DHP derivatives in the same preparation (13,15). A large number of DHP receptor sites have been found in the brain and they have physiological roles in the regulation of cardiovascular activities. Calcium channel blockers may produce an excitatory effect of the nucleus tractus solitarius when administered centrally, resulting in hypotension and bradycardia in rats (15). Considering that the hydrophobic parameter ( the ratio value of hydrophobic surface area of the molecule to the hydrophillic) of CPU- 23 is close to that of nifedipine (0.546 vs 0.508) (16), it is reasonable to infer that CPU-23 may pass through the blood-brain barer to affect central regulation of cardiovascular activities as do the DHP derivatives. Interestingly, tetrandrine at the effective i.v. concentration after i.v. injection, did not cause significant cardiovascular effects after i.c.v. injection (10), suggesting that CPU-23 may be advantageous over tetrandrine in the treatment of hypertension.
Antiarrhythmic and Anti-ischaemic ActionsCalcium channel blockers have been used in the treatments of arrhythmia, especially supraventricular antiarrhythmia, with good clinical results (17). As noted above, CPU-23 inhibits cytosolic Ca2+ increase in ventricular myocytes, the pacemaker action potentials in sinoatrial node and the slow action potential of the papillary muscle in vitro. CPU-23 also exerts a bradycardia and has more pronounced effects on ECG of hypertensive rats than those of normotensive rats in vivo(4). All of these cardiac effects of CPU-23 in vitro and in vivo suggest that it may offer antiarrhythmic action. Actually, CPU-23 dose-dependently reduced the total number of ectopic beats and both the duration and the incidence of ventricular tachycardia (VT) and ventricular fibrillation (VF) occurring in the first 30 min following coronary artery ligation of rats. CPU-23 also reduced mortality due to ischemia reperfusion in a dose- dependent manner. At 2.5-5 mg kg-1 ( i.v.), CPU-23 abolished completely VF, which was accompanied by 100% survival (Table 4). These findings indicate that CPU-23 exerts antiarrhythmic and anti-ischaemic effects like other calcium channel blockers (7).
Table 4. Effets of intravenous CPU-23 on early post-infarction arrhymias and mortality in pentobarbitone-anaesthetized rats.
________________________________________________________________________
Total numbers VT VF Mortality
Group of ectopic beats _____________ ____________ (%)
n Dura. Inci. Dura. Inci.
________________________________________________________________________
Saline 10 915�197 93�22 100 53�27 70 20
CPU-23(mg/kg)
1 8 264�130** 37�26 63 2�2** 13* 13
2.5 6 40�18** 2�2** 33** 0** 0** 0**
5 8 3�1** 2�2** 13** 0** 0** 0**
________________________________________________________________________VT: ventricular tachycardia; VF: ventricular fibrillation. Dura.:duration (s), Inci.:incidence (%). Mortality (% in 30 min). Data shown are the mean � s.e.mean of (n) animals. *P< 0.05; **P< 0.01 when compared with control.
Hamodynamic Studies
When administered to anaesthetized SD rats, CPU-23 (2.5-5 mg kg-1, i.v.) produced a dose-dependent decrease in left ventricular pressure (LVP), their first derivative over time (dP/dtmax), pulsatile pressure (BP) and heart rate (HR). The above hamodynamic effects appeared quickly and the peak effects were reached within 30 seconds after drug administration. At 5 mg kg-1 , CPU-23 decreased the systolic blood pressure (SBP) by 39%, diastolic blood pressure (DBP) by 41%, LVP by 30%, +dP/dtmax by 38%, -dP/dtmax by 46% and HR by 20%. Most of the hemodynamic parameters recovered in 30 min, but HR and DBP remained significantly depressed even at 60 min after drug administration (9). To systematically compare the hamodynamic effects of CPU-23 with classical calcium channel blockers, nifedipine, verapamil and diltiazem were selected as standards and administered at doses which produced a similar hypotensive effect, namely nifedipine 0.3 mg kg-1, verapamil 1 mg kg-1 ,diltiazem 2 mg kg-1 and CPU-23 5 mg kg-1. At those doses, the rank order of effect in decreasing LVP and dP/dtmax is verapamil > CPU-23 = diltialzem > nifedipine, but in lowering HR is CPU-23 = verapamil = diltilzem > nifedipine (9). In ischaemic arrhythmic rats, we observed that although coronary artery ligation significantly lowered the arterial blood pressure, it did not further reduce the MAP in rats pretreated with CPU-23 at doses of 2.5-5 mg kg-1(7). It is suggested, by the fact that CPU-23 does not further reduce MAP after coronary artery ligation at doses that produced significant antiarrhythmic action and reduced MAP of normal rats, that CPU-23 may be beneficial to patients with cardiac arrhythmias, as arterial blood pressure would not drop further with the use of the drug during myocardial ischaemia.
SUMMARY
The results of our studies conducted at the molecular, cellular, tissue, organ and whole animal levels suggested that CPU-23 is a novel Ca2+ and delayed rectifier K+ channels blocker. Although derived from tetrandrine, CPU-23 interacts at distinct binding sites on L-type calcium channels, blocks Ca2+ and K+ currents and exerts a more potent vasorelaxation than tetrandrine. These data suggest that CPU-23, because of its unique chemical structure, may be a useful tool for the study of L-type Ca2+ and delayed rectifier K+ channels. From its therapeutic potentials of antihypertension and antiarrhythmics, it can be deduced that CPU-23 may be a useful therapeutic agent worthy of further study and development. One weakness of CPU-23 is that its potency and selectivity for the vasculature is less than dihydropyridine derivatives, but this may be improved by a better understanding of the structure-activity relationship of tetrahydroisoquinolines and their interaction with ion channels on vascular smooth muscle cells.
ACKNOWLEDGMENTS: This work was supported in large part by a grant from Hong Kong Jockey Club (Charities) Ltd. and partly by the Alberta Heritage Foundation for Medical Research. We thank Prof. S.X. Peng in Chinese Pharmaceutical University for the kind supply of tetrahydroisoquinolines.
REFERENCES
1. Gao Y, Chang MY, Mao HY, Chen DH. The clinical observation of tetrandrine in the treatment of 270 cases of hypertension patients and hypertensive crisis. J. Chinese Int. Med. 1965;13:504-507.
2. Liu QY, Li B, Gang GM, Karpinski E, Pang PKT. Tetrandrine, a Ca++ antagonist: effects and mechanisms of action in vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 1995;273:32-39.
3. King VF, Garcia ML, Himmel D, Reuben JP, Lam YKT, Pan JX, Han GO, Kaczorowski GJ. Interaction of tetrandrine with slowly inactivating calcium channels: characterisation of calcium channel modulation by an alkaloid of Chinese medicinal herb origin. J. Biol. Chem. 1988;263:2238-2244.
4. Dong H, Waldron GJ, Cole WC, Triggle CR. Cardiovascular pharmacology of CPU-23: a novel calcium channel blocker. Cardiovasc. Drug Rev. 1996; 14 (4):364-379.
5. Dong H. Trends in calcium antagonists and their receptors. Fudan Lectures in Neurobiology. 1994; X:37-50.
6. Dong H, Lee CM, Huang WL, Peng SX. Cardiovascular effects of substituted tetrahydroisoquinalines in rats. Br. J. Pharmacol. 1992;107:262-268
7. Dong H, Sheng JZ, Lee CM, Wong TM. Calcium antagonistic and antiarrhythmic action of CPU-23, a substituted tetrahydroisoquinaline. Br. J. Pharmacol. 1993;109:113-119.
8. Depover A, Matlib MA, Lee SW, Dube GP, Grupp IL, Grupp G, Schwartz A. Specific binding of [3H]-nitrendipine to membrane from coronary arteries and heart in relation to pharmacological effects. Paradoxical stimulation by diltiazem. Biochem. Biophys. Res. Commun. 1982;108:110-117.
9. Dong H, Earle ML, Jiang YF, Loutzenhiser KA, Triggle CR. Cardiovascular effects of CPU-23, a novel L-type calcium channel blocker with a unique molecular structure. Br. J. Pharmacol. 1997; 122:1271-1278.
10. Qian JQ, Thoolen MJMC, van Mell JCA, Timmermans PBMWM, van Zwietan PA. Hypotensive activity of tetrandrine in rats: investigation into its mode of action. Pharmacology. 1983;26:187-197.
11. Todd GL, Sterns DA, Plambeck RD, Joekel CS, Eliot R. Protective effects of slow calcium channel antagonists on noradrenaline induced myocardial necrosis. Cardiovasc. Res. 1986;20:645-651.
12. Briand V, Laurent S, Tsoucaris-Kupfer D, Legrand M, Brisac AM, Schmitt H. Central and periphearl cardiovascular effects of the enantiomers of the calcium antagonist PN 200-110. Eur. J. Pharmacol. 1988;150:43-50.
13. Brisac AM, Huguet F, Champeroux P, Montastruc JL, Lucet B, Gerard P, Laurent S, Narcisse G, Schmitt H. Central interactions between dihydropyridines and cholinergic system in the control of blood pressure in rat. Brain Res. 1987;435:160-166.
14. Dong H, Lee CM, Ng KW, Wong TM. Central Cardiovascular Effects of CPU-23, a substituted tetrahydroisoquinaline, in Rats. Arch. int. Pharmacodyn. 1995;329:245-254.
15. Higuchi S, Takeshita A, Nadoya I, Tsutomu I, Hidayo M, Motoomi N. Arterial pressure and heart rate response to calcium channel blockers administered in the rat brain stem. Circ. Res. 1985;57:244-251.
16. Xu GY, Wei BY, Hua WY, Peng SX. Studies on molecular modelling of compound CPU- 23 and nifedipine and their comparative analysis. Proceedings of the third China-Japan drug design and development symposium. 1993;43-45.
17. Vanhoutte PM, Paoletti R. The WHO classification of calcium antagonists. Trends Pharmacol. Sci. 1987;8:4-5.
| Discussion Board | Previous Page | Your Symposium |