Invited Symposium: Intracellular Traffic of Organelles
| INABIS '98 Home Page | Your Session | Symposia & Poster Sessions | Plenary Sessions | Exhibitors' Foyer | Personal Itinerary | New Search |
Introduction
In fat and skeletal muscle insulin induces a rapid stimulation in cellular glucose uptake (for review see [1]). This acute activation in glucose transport is dependent on the recruitment of the insulin-regulated glucose transporter, GLUT4, from specialised intracellular storage vesicles to the plasma membrane [2]. The translocation of GLUT4 containing vesicles to the cell surface in both these tissues probably represents one of the best known examples of regulated intracellular organelle traffic. Surprisingly, however, we still know very little about (i) the mechanism by which insulin triggers the translocation of GLUT4, (ii) the nature of the intracellular GLUT4 pool and (iii) the mechanism by which GLUT4 vesicles are targeted and fused with the plasma membrane. This presentation discusses some recent work from our group and that of others which has attempted to address some of the pertinent issues related to these three areas.
Early signalling molecules
Early signalling molecules involved in the insulin-induced translocation of GLUT4 vesicles - the role of PI3K and Protein Kinase B (PKB/Akt)
The signalling process is initiated by the activated insulin receptor kinase which tyrosine phosphorylates a number of key intracellular target substrates, in particular IRS-1 (insulin receptor substrate 1) and its closely related relatives IRS-2 and IRS-3 [3-6]. Of the numerous IRS binding proteins, the most likely candidate involved in the hormonal activation of glucose transport is the serine/lipid kinase phosphoinositide 3-kinase (PI3K) [7-10]. The use of two structurally unrelated compounds, wortmannin and LY294002, which potently inhibit PI3K, have revealed that it plays a crucial role in regulating insulin stimulated GLUT4 translocation and glucose transport [11-14]. Precisely how PI3K signals onto the GLUT4 storage pool remains poorly understood, but it is unlikely to involve either MAP kinase or p70s6 kinase (which lie downstream of PI3K) since neither PD 98059 nor rapamycin, which respectively block activation of these kinases, inhibit insulin-stimulated glucose uptake [15]. The possibility that PI3K may directly regulate GLUT4 translocation has also been suggested since insulin stimulates its activity in microsomal membranes enriched with GLUT4 in fat cells [10,16]. Interestingly, one study has reported that IRS-1/PI3K complexes are targeted directly onto GLUT4 vesicles following insulin treatment [17]. Whilst this notion is clearly attractive this finding remains unsubstantiated and the view that other, as yet unidentified, signalling molecules participate in the translocation of GLUT4 remains a more likely possibility. This supposition has been strengthened by the demonstration that Protein Kinase B (PKB), which also lies downstream of PI3K, is involved in mediating insulin�s effects on glucose transport and glycogen metabolism [18-21].
PKB is rapidly activated by insulin in a PI3K-dependent manner [22-24]. This activation relies upon the phosphorylation of two key amino acid residues in PKB, Thr308 and Ser473, with full activation requiring phosphorylation of both [24]. The N-terminal domain of PKB contains a pleckstrin homology (PH) domain thought to be critical in allowing the kinase to interact with phospholipids and possibly other signalling molecules [25-27]. PKB activation is preceded by its recruitment to the PM [28]. How this occurs is unclear, but it has been suggested that the binding of PtdIns(3,4,5)P3 or PtdIns (3,4) P2 to the PH domain of PKB may be important in inducing conformational changes which enable it to become activated by two upstream kinases. Indeed, two recent reports have identified a 3�-phosphoinositide-dependent kinase (PDK1) which phosphorylates Thr308 and partially activates PKB [29,30]. PDK1 was recently cloned [31], but given that full activation requires phosphorylation of Ser473 , it is likely that another, as yet unidentified, upstream kinase (PDK2?) participates in PKB activation [25,32].
Glycogen synthase kinase-3 [20,21] and the cardiac isoform of 6 phosphofructo-2-kinase [33,34] represent to known physiological targets for PKB, but more recently its involvement in the regulation of glucose transport has also been highlighted. We have recently found that over expression of a constitutively active form of PKB in L6 muscle cells results in a significant stimulation in glucose uptake, at least to a level comparable to that elicited by insulin in control cells. This upregulation in glucose uptake is attributable to the increased translocation of GLUT4 to the PM (Figure 1) [35], an observation that has also been made in both 3T3-L1 and primary rat adipocytes expressing constitutively active forms of PKB [18,19]. However, recent work from Kasuga�s lab has reported that expression of a putative dominant negative form of PKB, whilst effective in blocking insulin-stimulated protein synthesis, does not inhibit the translocation of GLUT4 in 3T3-L1 adipocytes [36]. The reason for this apparent discrepancy is unclear, but we have recently suggested that only a small activation in PKB (~3-fold) may be required to induce a full increase in glucose uptake in L6 cells. Thus, if the activity of the endogenous PKB is not totally suppressed by over-expressing the dominant negative PKB then one may still observe an increase in insulin-stimulated glucose uptake [35]. At present no specific inhibitors of PKB exist, but data from three different insulin responsive cell types (L6 myotubes, 3T3-L1 and primary rat adipocytes) collectively support the notion that PKB plays an important role in the hormonal induced translocation of GLUT4. Moreover, by implication, we would argue that the upstream kinase, PDK1, is also likely to be a key component in the signal transduction process that initiates GLUT4 vesicle movement.
 Fig.1: Effect of a constitutive active PKB overexpressed in L6 myotubes on glucose transport and GLUT4 translocation.
Fig.1: Effect of a constitutive active PKB overexpressed in L6 myotubes on glucose transport and GLUT4 translocation.
Intracellular GLUT4 compartments
Intracellular GLUT4 compartments in insulin-sensitive tissues
Precisely how the signal from the insulin-receptor is routed to the intracellular GLUT4 compartment remains poorly understood although, as indicated above, it has been reported that PI3K [17], and more recently PKB [37], may associate directly with the intracellular GLUT4 pool in adipocytes. This suggestion would imply that GLUT4 vesicles contain specific proteins that may couple or transduce the hormonal signal into one that initiates the physical movement of the vesicle to the cell surface. In order to identify such proteins a number of labs have attempted to gain insights into the nature of the intracellular GLUT4 pool in fat and skeletal muscle (for review see [2]).
Use of impermeant photolabels that bind GLUTs have suggested that, even in the absence of any hormonal treatment, GLUT4 recycles constitutively between the cell surface and an intracellular compartment [38]. However, a single intracellular GLUT4 pool is likely to be a gross over-simplification since immunogold staining of unstimulated adipocytes has shown that most of the cellular GLUT4 can be localised to as many as eleven different intracellular locations resembling what appear as tubulo-vesicular structures close to the trans-Golgi network region [39]. Upon insulin treatment approximately half of the immunogold labelling seen in these sites is lost, presumably to the surface membrane [39]. Moreover, work from Holman�s group has suggested, based on mathematical analyses and kinetic modelling, that two, rather than just one, intracellular GLUT4 pools best describe the observed rate of activation of glucose transport and GLUT4 recycling in the presence of insulin [40]. Indeed, biochemical evidence for the existence of more than one GLUT4 pool has also been documented. Livingstone et al have shown, using a technique called compartment ablation, that up to 40% of the intracellular GLUT4 in 3T3-L1 adipocytes is present within a compartment that also contains the transferrin receptor (TfR) [41]. Since TfR is thought to be a good marker of the recycling endosomes, the GLUT4 resident within this pool is considered to represent that which constitutively recycles between the plasma membrane and early endosomes. In contrast, nearly 60% of the intracellular GLUT4 was localised within a pool that could not be ablated and which did not contain any Tfr [41].
In skeletal muscle, the idea that there may be more than one intracellular GLUT4 pool has been established for a considerably longer period of time. This long-standing view is based on the finding that in addition to insulin, glucose transport can also be stimulated in response to muscle exercise or contraction and that the effects of insulin and exercise are fully additive (for review see [42]). The stimulatory effects of exercise on glucose uptake, like insulin, also involve the increased recruitment of intracellular GLUT4 to the sarcolemma [42-44]. Very little information exists concerning the identity of the intracellular pool that donates GLUT4 in response to insulin or muscle contraction, but it is thought that both stimuli �tap� different GLUT4 compartments and do so by distinct signalling mechanisms given that insulin-response is blocked by the PI3K inhibitor, wortmannin, whereas the inhibitor has no effect on the contraction-mediated response [8,42,45,46].
In an attempt to gain some insight into the nature of intracellular GLUT4 pools present in skeletal muscle we decided to immunoisolate intracellular GLUT4 vesicles from rat skeletal muscle. In brief, we used a well established subcellular fractionation procedure [47,48] to isolate crude muscle membranes which were subsequently fractionated on a discontinuous sucrose gradient (25%, 30% and 35% sucrose w/w). Membranes recovered from the top of each sucrose interface were removed and denoted F25, F30 and F35, respectively. Previous work has shown that membranes recovered in the F25 are ostensibly sarcolemmal in origin, based on the immunological enrichment in this fraction of plasma membrane (PM) markers (i.e. the 1 subunit of the Na,K-ATPase and GLUT1, Figure 2). Membranes from the F30 had not previously been characterised to any significant extent, whereas those recovered in the F35 have been shown to house the insulin-responsive GLUT4 pool [47,48]. In line with this view, Figure 2 shows that when the three membrane fractions were prepared from control and insulin-treated muscle the abundance of GLUT4 fell in the F35 fraction prepared from insulin-treated muscle. This loss in GLUT4 was recovered in the F25 consistent with the idea that GLUT4 had translocated from an intracellular membrane pool to the sarcolemma. Interestingly, the F30 harbours approximately the same amount of GLUT4 as the F35 (when the total protein recoveries of the two fractions are taken into account), but does not show any insulin-dependent change in GLUT4 content. The F30 is not contaminated to any significant extent with sarcolemma (based on the lack of any 1-Na,K-ATPase subunits in this fraction), but is enriched with Tfr tending to signify that whilst intracellular this fraction may largely be of endosomal origin (Figure 2). Membranes in the F30 are likely to be different from those in the F35 based on the finding that Tfr was not detected in this fraction (Figure 2).
Since both F30 and F35 contain a significant pool of GLUT4 we asked the question of whether these fractions contained distinct populations of GLUT4 vesicles. This possibility was addressed by immunoprecipitating GLUT4 vesicles from the F30 and F35 using established methodology [47,48] and screening them with antibodies to proteins of interest. Figure 2 shows an analyses of GLUT4 vesicles which were immunoprecipitated from both fractions with an efficiency approaching ~80% [48]. TfR and the insulin regulated aminopeptidase, vp165 (also known as IRAP), were coprecipitated with GLUT4 vesicles isolated from the F30, whereas only vp165 was present in vesicles isolated from the F35 (Figure 2). This finding implies that GLUT4 vesicles from the F30, which contain TfR, may represent an endosomal pool that constitutively recycles at the cell surface. In contrast, GLUT4 vesicles from the F35 may be regarded as the insulin-responsive �storage pool� based on the observation that this pool shows a net loss in GLUT4 following insulin treatment and lacks TfR. This proposition is further strengthened by the finding that GLUT4 vesicles from the F35 also contain the small monomeric G protein Rab4 [49], which has been implicated in the insulin regulated movement of GLUT4 in rat adipocytes [50]. Our findings are also in good agreement with those recently reported by Zorzano�s group who have also reported the presence in skeletal muscle of two distinct intracellular GLUT4 compartments; one which responds to insulin and the other which does not [51]. Furthermore, our data is broadly consistent with recent electron microscopic data showing that GLUT4 can be sub-divided into two types of clusters; TfR negative elements which are insulin responsive and TfR positive elements which are recruited to the sarcolemma in response to muscle contraction [52]. However, it is highly unlikely that the TfR/GLUT4 positive pool observed in our studies (i.e. that contained in the F30) represents the exercise-responsive pool given that previous work has shown that exercise does not induce any significant loss in glucose transporters from the F30 fraction [44]. The precise role of the TfR/GLUT4 pool in our studies therefore remains currently unknown, but assessing whether the insulin-responsive pool donates its GLUT4 complement directly to the sarcolemma or if GLUT4 transporters are routed via the endosomal (TfR positive) pool remains an interesting topic for future study.
 Fig.2:A: Subcellular distribution of different proteins in rat skeletal muscle fractions prepared by sucrose-gradient fractionation. B: Effects of insulin on the subcellular distribution of GLUT4 in muscle membrane fractions. C: Analyses of the co-localisation of vp165, TfR and cellubrevin with GLUT4 vesicles isolated from the F30 and F35 internal fractions of rat skeletal muscle.
Fig.2:A: Subcellular distribution of different proteins in rat skeletal muscle fractions prepared by sucrose-gradient fractionation. B: Effects of insulin on the subcellular distribution of GLUT4 in muscle membrane fractions. C: Analyses of the co-localisation of vp165, TfR and cellubrevin with GLUT4 vesicles isolated from the F30 and F35 internal fractions of rat skeletal muscle.
Targeting and fusion of GLUT4 vesicles
Targeting and fusion of GLUT4 vesicles - the role of SNAREs
Over the past few years there has been significant interest in trying to define the mechanism by which GLUT4 vesicles may be targeted and fused with the plasma membrane of fat cells and skeletal muscle. In particular, considerable focus has been placed on the idea that the targeting and fusion of GLUT4 vesicles may resemble regulated neurosecretion given that proteins implicated in synaptic vesicle fusion have also been shown to be expressed in insulin-sensitive tissues. Our current understanding of synaptic vesicle fusion is based on the model put forward by Rothman and colleagues [53] now widely known as the SNARE (SNAP receptor) hypothesis. This model proposes that fusion of synaptic vesicles with the pre-synaptic membrane is directed by the formation of a SNARE complex that is generated by the association of a specific vesicle associated protein (termed a vSNARE) with an appropriate target SNARE (tSNARE) resident in the plasma membrane. Examples of vSNAREs include the synaptobrevins VAMP (vesicle associated membrane protein) and cellubrevin and tSNAREs such as synaptosome-associated protein (SNAP)-25 and syntaxin. The formation of the v-t-SNARE scaffold then allows for the binding of two additional cytosolic proteins, NSF and -SNAP. NSF is an ATPase and hydrolysis of bound ATP is thought to provide the energy for the membrane fusion event.
So what experimental evidence exists implicating SNAREs in GLUT4 translocation? In addition to data showing that vSNAREs (VAMP2 and cellubrevin) and tSNAREs (syntaxin4 and SNAP23) are expressed in insulin-sensitive tissues evidence also exists showing that GLUT4 vesicles isolated from adipocytes harbour VAMP2 and cellubrevin [54-57]. The evidence for the functional involvement of SNARE proteins in GLUT4 translocation however has largely, if not exclusively, been obtained from studies in the murine 3T3-L1 adipocytes. Both VAMP2 and cellubrevin are proteolytic targets for clostridial toxins (i.e. botulinum and tetanus toxins) and their introduction into permeabilised 3T3-L1 adipocytes has been shown to reduce GLUT4 translocation [57,58]. Moreover, the introduction of recombinant syntaxin4 or anti-syntaxin4 antibodies into 3T3-L1 adipocytes also impairs GLUT4 translocation thereby providing further support for the notion that these proteins may participate in insulin regulated GLUT4 traffic [59-61].
So does the �SNARE phenomena� which has been so firmly established in 3T3-L1 adipocytes extend to isolated rat adipocytes and skeletal muscle? We argued that if the presence of these v-SNAREs was critical for the insulin-induced movement and fusion of GLUT4 vesicles, then their ablation should also block any increase in cell surface GLUT4 and glucose transport elicited by insulin in isolated rat adipocytes. As with 3T3-L1 adipocytes, regular fat cells do not possess the receptors that can bind clostridial toxins. To overcome this difficulty we decided to introduce tetanus toxin into rat adipocytes by electroporation; a procedure that transiently creates pores in the membrane enabling large proteins, such as the toxin, to enter into the cell. Once internalised the toxin can cleave its protein substrates i.e. the vSNAREs. What our study showed was that we were able to successfully ablate the expression of cellubrevin and VAMP2 following adipocyte electroporation in the presence of tetanus toxin [62]. However, despite the proteolysis of both VAMP and cellubrevin we found that insulin was still capable of stimulating GLUT4 translocation and glucose transport in these cells (Figure 3). The reasons for the apparent discrepancy between our data and that obtained using 3T3-L1 adipocytes remains unclear, but it should be stressed that, to our knowledge, no study has shown a complete loss in insulin-stimulated GLUT4 translocation following SNARE manipulation. Indeed, in most cases the inhibitory effect of toxins, recombinant SNARE proteins or antibodies on GLUT4 translocation and glucose transport is highly variable (from between 15 to 80%). However, since we find that both cellubrevin and VAMP2 are resident in GLUT4 vesicles isolated from rat adipocytes [62] we believe that they must fulfil some role related to the fusion of these vesicles. Given that there is growing acceptance of the idea that more than one intracellular GLUT4 pool exists in muscle and fat, it is possible that VAMP2 and cellubrevin regulate fusion of GLUT4 vesicles between different intracellular compartments and that fusion with the plasma membrane may involve another, as yet unidentified, vSNARE. We are currently assessing this possibility.
The role of SNAREs in muscle GLUT4 translocation in skeletal muscle is far from clear although some investigators have been happy to take the view that what has reportedly been documented in 3T3-L1 adipocytes also holds for muscle. This assumption has partly been based on the finding that skeletal muscle expresses VAMP2, cellubrevin and the tSNAREs syntaxin 4 and SNAP23 [63]. We, like others, also find that VAMP2 and cellubrevin are expressed in skeletal muscle [64], but in our hands, analyses of GLUT4 vesicles isolated from the F30 and the insulin-responsive muscle fraction (i.e. the F35) reveals that neither VAMP2 or cellubrevin can be coprecipitated with GLUT4 [64] (Figure 2). Both proteins were recovered in the immunesupernatant under conditions when ~80% of the GLUT4 vesicles had been precipitated. These observations imply that whilst ~20% of the vesicles are not isolated a substantial population of GLUT4 vesicles do not possess cellubrevin or VAMP2. Work from Zorzano�s group has shown that GLUT4 vesicles isolated from muscle fractions generated using a different experimental approach to us does enable the detection of cellubrevin and VAMP2 in distinct intracellular GLUT4 pools. However, the amount of each vSNARE coprecipitated was found to be very small (<20% of the total expressed in the fraction) [65]. The finding that cellubrevin and VAMP2 are expressed in some, but not all, GLUT4 compartments implies that if these proteins are involved in membrane fusion then they may perform distinct roles in those compartments in which they are resident. Interestingly, a similar type of analyses of GLUT4 vesicles isolated from cardiomyocytes reveals that they lack VAMP2; an observation that signifies that GLUT4 vesicles from skeletal muscle and fat are compositionally different to those found in heart [65]. Thus the question of how important the SNARE hypothesis is with respect to GLUT4 translocation in skeletal muscle remains very much open in our view. We are not aware of any studies that have attempted to address the functional involvement of cellubrevin and VAMP2 (or that of the tSNAREs) in the insulin-regulated translocation of GLUT4 in skeletal muscle. If cellubrevin and VAMP2 are not involved then it is likely that the targeting and fusion of GLUT4 vesicles in muscle either relies upon other SNARE proteins which have yet to be identified or occurs by a mechanism that is fundamentally different to that operating in fat cells.
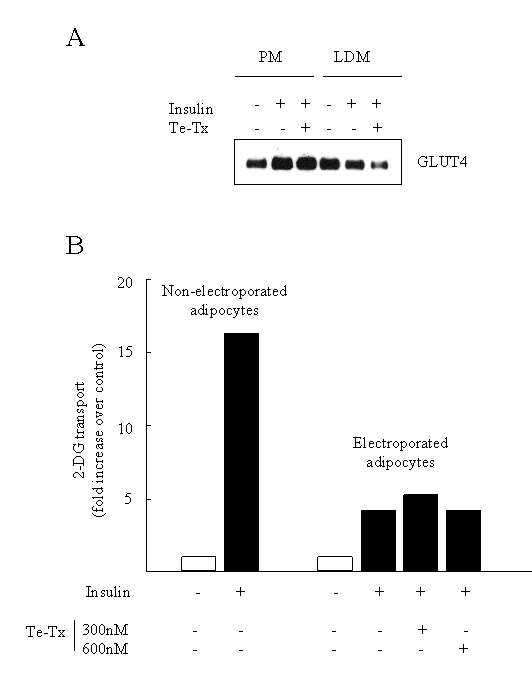 Fig.3: A: Representative Western blot showing the effects of insulin and Te-Tx on the subcellular distribution of GLUT4 in electroporated adipocytes. B: Effects of insulin on glucose transport in rat adipocytes following electroporation with Te-Tx .
Fig.3: A: Representative Western blot showing the effects of insulin and Te-Tx on the subcellular distribution of GLUT4 in electroporated adipocytes. B: Effects of insulin on glucose transport in rat adipocytes following electroporation with Te-Tx .
References
We are grateful to Carlos Aledo and Froogh Darakhshan who participated in some of the studies reported. Work in our lab is supported by the Wellcome Trust, British Diabetic Association and BBSRC.
- Holman GD, Kasuga M (1997) From receptor to transporter: insulin signalling to glucose transport. Diabetologia 40: 991-1003
- Stephens JM, Pilch PF (1995) The metabolic-regulation and vesicular transport of GLUT4, the major insulin-responsive glucose-transporter. Endocrine Reviews 16: 529-546
- White MF, Kahn CR (1994) The insulin signaling system. J Biol Chem 269: 1-4
- Araki E, Lipes MA, Patti ME et al. (1994) Alternative pathway of insulin signaling in mice with targeted disruption of the IRS-1 gene. Nature 372: 186-190
- Hansen PA, Corbett JA, Holloszy JO (1997) Phorbol esters stimulate muscle glucose transport by a mechanism distinct from the insulin and hypoxia pathways. Am J Physiol 36: E28-E36
- Kaburagi Y, Satoh S, Tamemoto H et al. (1997) Role of insulin receptor substrate-1 and pp60 in the regulation of insulin-induced glucose transport and GLUT4 translocation in primary adipocytes. J Biol Chem 272: 25839-25844
- Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR (1994) Phosphatylinositol 3-kinase activation is required for insulin stimulation off pp70 s6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol 14: 4902-4911
- Tsakiridis T, McDowell HE, Walker T et al. (1995) Multiple roles of phosphatidylinositol 3-kinase in regulation of glucose-transport, amino-acid-transport, and glucose transporters in L6 skeletal-muscle cells. Endocrinology 136: 4315-4322
- Quon MJ, Chen H, Ing BL et al. (1995) Roles of 1-phosphatidylinositol 3-kinase and ras in regulating translocation of GLUT4 in transfected rat adipose-cells. Mol Cel Biol 15: 5403-5411
- Yang J, Clarke JF, Ester CJ, Young PW, Kasuga M, Holman GD (1996) Phosphatidylinositol 3-kinase acts at an intracellular membrane site to enhance GLUT4 exocytosis in 3T3-L1 cells. Biochem J 313: 125-131
- Kanai F, Ito K, Todaka M et al. (1993) Insulin-stimulated GLUT4 translocation is relevant to the phosphorylation of IRS-1 and the activity of PI3-Kinase. Biochem Biophys Res Com 195: 762-768
- Clarke JF, Young PW, Yonezawa K, Kasuga M, Holman GD (1994) Inhibition of the translocation of GLUT1 and GLUT4 in 3t3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem J 300: 631-635
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M (1994) Essential role of phosphatylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. J Biol Chem 269: 3568-3573
- McDowell HE, Walker T, Hajduch E et al. (1997) Inositol phospholipid 3-kinase is activated by cellular stress but is not required for the stress-induced activation of glucose transport in L6 rat skeletal muscle cells.. Eur J Biochem 247: 306-313
- McDowell HE, Walker T, Downes CP, Hundal HS: 1994 Inhibition of IGF-1 stimulated glucose and System A amino acid transport in L6 rat skeletal muscle cells: effects of wortmannin (WM) and Rapamycin (RAP). [Abstract] J Physiol 482: 5P
- Nave BT, Haigh RJ, Hayward AC, Siddle K, Shepherd P (1996) Compartment-specific regulation of phosphoinositide 3-kinase by platelet-derived growth-factor and insulin in 3T3-L1 adipocytes. Biochem J 318: 55-60
- Heller-Harrison RA, Morin M, Guilherme A, Czech MP (1996) Insulin-mediated targeting of phosphatidylinositol 3-kinase to GLUT4- containing vesicles. J Biol Chem 271: 10200-10204
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA (1996) Expression of a constitutively active Akt ser/thr kinase in 3T3- L1 adipocytes stimulates glucose-uptake and glucose-transporter-4 translocation. J Biol Chem 271: 31372-31378
- Tanti JF, Grillo S, Gremeaux T, Coffer PJ, Vanobberghen E, Lemarchandbrustel Y (1997) Potential role of Protein Kinase B in glucose transporter 4 translocation in adipocytes. Endocrinology 138: 2005-2010
- Cohen P, Alessi DR, Cross DAE (1997) The tenth datta lecture - PDK1, one of the missing links in insulin signal transduction? FEBS Letters 410: 3-10
- Shaw M, Cohen P, Alessi DR (1997) Further evidence that the inhibition of glycogen synthase kinase- 3 beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser- 9 and not by dephosphorylation of Tyr-216. FEBS Letters 416: 307-311
- Cross DAE, Alessi DR, Cohen P, Andjelkovic M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin-mediated protein kinase B. Nature 378: 785-789
- Moule SK, Welsh GI, Edgell NJ, Foulstone EJ, Proud CG, Denton RM (1997) Regulation of Protein Kinase B and glycogen synthase kinase-3 by insulin and beta-adrenergic agonists in rat epididymal fat cells: Activation of Protein Kinase B by wortmannin-sensitive and - insensitive mechanisms. J Biol Chem 272: 7713-7719
- Alessi DR, Andjelkovic M, Caudwell B et al. (1996) Mechanism of activation of Protein Kinase B by insulin and IGF-1. EMBO J 15: 6541-6551
- Hemmings BA (1997) Update: Signal transduction - PtdIns(3,4,5)P-3 gets its message across. Science 277534
- Coffer PJ, Woodgett JR (1991) Molecular-cloning and characterization of a novel putative protein- serine kinase related to the cAMP-dependent and Protein-Kinase-C families. Eur J Biochem 201: 475-481
- Datta K, Franke TF, Chan TO et al. (1995) AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol Cell Biol 15: 2304-2310
- Andjelkovic M, Alessi DR, Meier R et al. (1997) Role of translocation in the activation and function of Protein Kinase B. J Biol Chem 272: 31515-31524
- Alessi DR, James SR, Downes CP et al. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B. Curr Biol 7: 261-269
- Stokoe D, Stephens LR, Copeland T et al. (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of Protein Kinase B. Science 277: 567-570
- Alessi DR, Deak M, Casamayor C et al. (1997) 3-phosphoinositide dependent protein kinase-1 (PDK1); structural and functional homology with the drosophila DSTPK61 kinase. Curr Biol 7: 776-789
- Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387: 673-676
- Lefebvre V, Mechin MC, Louckx MP, Rider MH, Hue L (1996) Signaling pathway involved in the activation of heart 6-phosphofructo-2-kinase by insulin. J Biol Chem 271: 22289-22292
- Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH (1997) Phosphorylation and activation of heart 6-phosphofructo-2-kinase by Protein Kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 272: 17269-17275
- Hajduch E, Alessi DR, Hemmings BA, Hundal HS (1998) Constitutive activation of Protein Kinase B (PKB) by membrane targeting promotes glucose and System A amino acid transport, protein synthesis and GSK3 inactivation in L6 muscle cells. Diabetes 47: 1006-1013
- Kitamura T, Ogawa W, Sakaue H et al. (1998) Requirement for activation of the serine-threonine kinase akt (protein kinase B) in insulin stimulation of protein synthesis but not of glucose transport. Mol Cel Biol 18: 3708-3717
- Calera MR, Martinez C, Liu H, El Jack AK, Birnbaum MJ, Pilch PF (1998) Insulin increases the association of Akt-2 with GLUT4-containing vesicles. J Biol Chem 273: 7201-7204
- Satoh S, Nishimura H, Clark AE et al. (1993) Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose-cells -evidence that exocytosis is a critical site of hormone action. J Biol Chem 268: 17820-17829
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose-tissue of the rat. J Cell Biol 113: 123-135
- Holman GD, Leggio LL, Cushman SW (1994) Insulin-stimulated glut4 glucose-transporter recycling - a problem in membrane-protein subcellular trafficking through multiple pools. J Biol Chem 269: 17516-17524
- Livingstone C, James DE, Rice JE, Hanpeter D, Gould GW (1996) Compartment ablation analysis of the insulin responsive glucose transporter (GLUT4) in 3T3-L1 adipocytes. Biochem J 315: 487-495
- Hayashi T, Wojtaszewski JFP, Goodyear LJ (1997) Exercise regulation of glucose transport in skeletal muscle. Am J Physiol 273: E1039-E1051
- Douen AG, Ramlal T, Klip A, Young DA, Cartee GD, Holloszy JO (1989) Exercise-induced increase in glucose transporters in plasma membranes of rat skeletal muscle. Endocrinology 124: 449-454
- Douen AG, Ramlal T, Rastogi S et al. (1990) Exercise induces recruitment of the "insulin-responsive glucose transporter". J Biol Chem 265: 13427-13430
- Yang J, Clarke JF, Ester CJ, Young PW, Kasuga M, Holman GD (1996) Phosphatidylinositol 3-kinase acts at an intracellular membrane site to enhance GLUT4 exocytosis in 3T3-L1 cells. Biochem J 313: 125-131
- Lee AD, Hansen PA, Holloszy JO (1995) Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose-transport activity in skeletal-muscle. FEBS Letters 361: 51-54
- Marette A, Richardson JM, Ramlal T et al. (1992) Abundance, localization and insulin-induced translocation of glucose transporters in red and white muscle. Am J Physiol 263: C443-C452
- Aledo JC, Lavoie L, Volchuk A, Keller SR, Klip A, Hundal HS (1997) Identification and characterization of two distinct intracellular GLUT4 pools in rat skeletal muscle: evidence for an endosomal and an insulin-sensitive GLUT4 compartment. Biochem J 325: 727-732
- Aledo JC, Darakhshan F, Hundal HS (1995) Rab4, but not the transferrin receptor, is colocalized with GLUT4 in an insulin-sensitive intracellular compartment in rat skeletal-muscle. Biochem Biophys Res Com 215: 321-328
- Cormont M, Tanti J, Zahraoui A, Van Obberghen E, Tavitian A, Le Marchand-Brustel Y (1993) Insulin and okadaic acid induce Rab4 redistribution in adipocytes. J Biol Chem 268: 19491-19497
- Munoz P, Rosemblatt M, Testar X, Palacin M, Zorzano A (1995) Isolation and characterization of distinct domains of sarcolemma and t-tubules from rat skeletal-muscle. Biochem J 307: 273-280
- Ploug T, vanDeurs B, Ai H, Cushman SW, Ralston E (1998) Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol 142: 1429-1446
- Rothman JE (1994) Mechanisms of intracellular protein transport. Nature 372: 55-63
- Corley Cain C, Trimble WS, Lienhard GE (1992) Members of the VAMP family of synaptic vesicle proteins are components of glucose transporter-containing vesicles from rat adipocytes. J Biol Chem 267: 11681-11684
- Volchuk A, Sargeant R, Sumitani S, Liu Z, He L, Klip A (1995) Cellubrevin is a resident protein of insulin-sensitive GLUT4 glucose transporter vesicles in 3T3-L1 adipocytes. J Biol Chem 270: 8233-8240
- Martin S, Tellam J, Livingstone C, Slot JW, Gould GW, James DE (1996) The glucose-transporter (GLUT4) and vesicle-associated membrane protein-2 (VAMP-2) are segregated from recycling endosomes in insulin-sensitive cells. J Cell Biol 134: 625-635
- Tamori Y, Hashiramoto M, Araki S et al. (1996) Cleavage of vesicle-associated membrane protein (VAMP)-2 and Cellubrevin on GLUT4-containing vesicles inhibits the translocation of GLUT4 in 3T3-L1 adipocytes.. Biochem Biophys Res Commun 220: 740-745
- Macaulay SL, Rea S, Gough KH, Ward CW, James DE (1997) Botulinum e toxin light chain does not cleave SNAP-23 and only partially impairs insulin stimulation of GLUT4 translocation in 3T3- L1 cells. Biochem Biophys Res Com 237: 388-393
- Olson AL, Knight JB, Pessin JE (1997) Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle snap receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol Cel Biol 17: 2425-2435
- Cheatham B, Volchuk A, Kahn CR, Wang L, Rhodes CJ, Klip A (1996) Insulin-stimulated translocation of GLUT4 glucose transporters requires SNARE-complex proteins. Proc Nat Acad Sci USA 93: 15169-15173
- Volchuk A, Wang QH, Ewart HS et al. (1996) Syntaxin 4 in 3T3-L1 adipocytes: regulation by insulin and participation in insulin-dependent glucose transport. Mol Biol Cell 7: 1075-1082
- Hajduch E, Aledo JC, Watts C, Hundal HS (1997) Proteolytic cleavage of cellubrevin and vesicle-associated membrane protein (VAMP) by tetanus toxin does not impair insulin- stimulated glucose transport or GLUT4 translocation in rat adipocytes. Biochem J 321: 233-238
- Volchuk A, Mitsumoto Y, He L et al. (1994) Expression of vesicle-associated membrane protein 2 (VAMP-2)/synaptobrevin II and cellubrein in rat skeletal muscle and in a muscle cell line. Biochem J 304: 139-145
- Aledo JC, Hajduch E, Darakhshan F, Hundal HS (1996) Analyses of the colocalization of cellubrevin and the GLUT4 glucose transporter in insulin-responsive tissues. FEBS Letters 395: 211-216
- Sevilla L, Tomas E, Munoz P et al. (1997) Characterization of two distinct intracellular GLUT4 membrane populations in muscle fiber. differential protein composition and sensitivity to insulin. Endocrinology 138: 3006-3015
| Discussion Board | Previous Page | Your Symposium |