| INABIS '98 Home Page | Your Symposium | Related Symposia & Posters | Scientific Program | Exhibitors' Foyer | Personal Itinerary | New Search |
The
AT1 subtype of angiotensin II (AngII) receptors
mediates
most of the effects of this agonist in the healthy
adult cardiovascular
system [1, 2], thus we will review here new
advances in
AT1-mediated
signalling.
Protein sequence homologies indicate that
AT1
belongs to the vast family of receptors with
seven transmembrane
domains, a notion consistent with
experiments indicating that
AngII signals via heterotrimeric
G-proteins [3]. Phospholipase
C (PLC) is activated by AngII
within 5 s in vascular smooth muscle
cells (vsmc), thus
generating diacylglycerol (DG) and inositol
trisphosphate
(IP3), an activator of intracellular
calcium
mobilization [1, 2]. G-proteins usually activate the
ß isoform
of PLC [4]. Thus the discovery that
AngII activates
PLC![]() via
tyrosine phoshorylation, just
like a typical growth factor,
appeared paradoxical [5]. AngII
increased PLC
via
tyrosine phoshorylation, just
like a typical growth factor,
appeared paradoxical [5]. AngII
increased PLC![]() 1 phosphorylation 4.5
fold at 30 s in rat vsmc.
Electroporation of c-Src antibodies or preincubation
with the
tyrosine kinase inhibitors genistein or tyrphostin A
inhibited
AngII-induced PLC
1 phosphorylation 4.5
fold at 30 s in rat vsmc.
Electroporation of c-Src antibodies or preincubation
with the
tyrosine kinase inhibitors genistein or tyrphostin A
inhibited
AngII-induced PLC![]() phosphorylation, IP3
production and calcium mobilization [5-8].
AngII induced phosphorylation
of tyrosine 319 in the cytoplasmic
domain of AT1, thus
permitting binding of
PLC
phosphorylation, IP3
production and calcium mobilization [5-8].
AngII induced phosphorylation
of tyrosine 319 in the cytoplasmic
domain of AT1, thus
permitting binding of
PLC![]() via its C-terminal
SH2
(Src homology 2) domain [9]. Opposite results were obtained
in human
vsmc, in which anti PLCß1 antibodies
inhibited
AngII-induced IP3 generation. In this
system, neither
anti PLC
via its C-terminal
SH2
(Src homology 2) domain [9]. Opposite results were obtained
in human
vsmc, in which anti PLCß1 antibodies
inhibited
AngII-induced IP3 generation. In this
system, neither
anti PLC![]() 1 antibodies,
nor incubation with genistein
or herbimycin A had any effect on
IP3 formation or
the calcium signal [10]. These
discrepancies were reconciled in a recent
study in rat vsmc
which showed that both mechanisms sequentially contributed
to
AngII-induced IP3 production. Before 30 s,
IP3
generation was insensitive to genistein and was
inhibited by
anti PLCß1 antibodies. In contrast, after 30
s both genistein
and anti PLC
1 antibodies,
nor incubation with genistein
or herbimycin A had any effect on
IP3 formation or
the calcium signal [10]. These
discrepancies were reconciled in a recent
study in rat vsmc
which showed that both mechanisms sequentially contributed
to
AngII-induced IP3 production. Before 30 s,
IP3
generation was insensitive to genistein and was
inhibited by
anti PLCß1 antibodies. In contrast, after 30
s both genistein
and anti PLC![]() 1
antibodies reduced IP3
accumulation [11]. The
presence of PLCß1 in rat vsmc was
confirmed by RT-PCR and
western blotting [10, 11]. In additional
experiments, early
coupling of AT1 to G-proteins was
demonstrated by
inhibition of AngII-induced IP3 generation
at 15 s
with anti G
1
antibodies reduced IP3
accumulation [11]. The
presence of PLCß1 in rat vsmc was
confirmed by RT-PCR and
western blotting [10, 11]. In additional
experiments, early
coupling of AT1 to G-proteins was
demonstrated by
inhibition of AngII-induced IP3 generation
at 15 s
with anti G![]() q/11,
anti
G
q/11,
anti
G![]() 12 and
Gß
antibodies, as well as by overexpression of the
ß
12 and
Gß
antibodies, as well as by overexpression of the
ß![]() binding domain of an
adrenergic receptor kinase
[11]. These studies demonstrate that
AngII couples sequentially,
first to PLCß1, through
Gß
binding domain of an
adrenergic receptor kinase
[11]. These studies demonstrate that
AngII couples sequentially,
first to PLCß1, through
Gß![]() as
well as two
different G
as
well as two
different G![]() subunits
(Fig. 1,
left panel); and second to PLC
subunits
(Fig. 1,
left panel); and second to PLC![]() 1
(Fig.
1, middle panel).
1
(Fig.
1, middle panel).
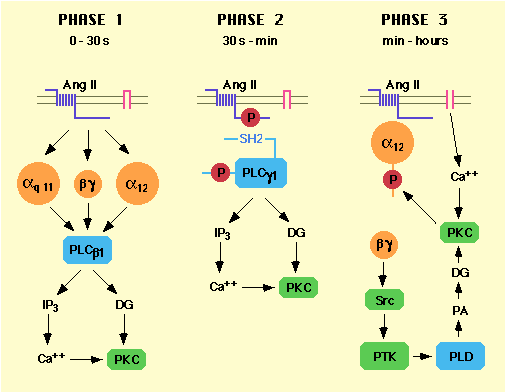
Figure 1. Sequential activation of different phospholipase subtypes by AngII in vsmc
Following transient generation of IP3
and
diacylglycerol (DG) by PLC (terminated within minutes), AngII
induced
a prolonged activation of phospholipase D (PLD) in vsmc
[12].
This resulted in a long-lasting accumulation of
phosphatidic
acid and its dephosphorylation product DG, that
accounted for
the sustained stimulation of protein kinase C
observed in vsmc
[2, 13]. Coupling of AT1 to PLD
activation was dependent
on several pathways in vsmc. The
heterotrimeric G protein activator
sodium fluoride almost
doubled PLD activity in vsmc. Furthermore,
AngII-induced PLD
activation was inhibited by 80% in cells overexpressing
a
ß![]() binding protein
and
more than 50% by electroporation of anti G
binding protein
and
more than 50% by electroporation of anti G![]() 12
or anti
Gß antibodies and 75% by anti-Src antibodies [14].
PLD activation
by AngII in vsmc also required extracellular calcium,
protein
kinase C [13, 15] and sequestration of AT1
in a
compartment such as caveolae [16]. Thus, initial activation
of
PLD presumably results from AT1-induced
dissociation
of G
12
or anti
Gß antibodies and 75% by anti-Src antibodies [14].
PLD activation
by AngII in vsmc also required extracellular calcium,
protein
kinase C [13, 15] and sequestration of AT1
in a
compartment such as caveolae [16]. Thus, initial activation
of
PLD presumably results from AT1-induced
dissociation
of G![]() 12 and ß
12 and ß![]() subunits which may activate PLD through a
tyrosine
kinase pathway. Sustained activation of PLD may result
from a
positive feedback loop in which protein kinase C,
stimulated
by calcium influx and PLD-generated DG,
phosphorylates G
subunits which may activate PLD through a
tyrosine
kinase pathway. Sustained activation of PLD may result
from a
positive feedback loop in which protein kinase C,
stimulated
by calcium influx and PLD-generated DG,
phosphorylates G![]() 12
[17], thus keeping ß
12
[17], thus keeping ß![]() subunits dissociated (Fig. 1, right panel).
These recent studies
have clarified the earliest coupling events important
for AngII
responses, leading to a greater understanding of AT1
coupling to
G proteins at the molecular level.
subunits dissociated (Fig. 1, right panel).
These recent studies
have clarified the earliest coupling events important
for AngII
responses, leading to a greater understanding of AT1
coupling to
G proteins at the molecular level.
Several
important AngII-induced signalling
cascades start with
activation of the Src family of tyrosine
kinases [18-21]. Src does
not bind to AT1, but may
be activated by
ß![]() subunits
of
G-proteins [14, 19]. In addition to activating PLC
subunits
of
G-proteins [14, 19]. In addition to activating PLC![]() and PLD, Src is probably responsible for
phosphorylation of
Shc in vsmc [18] as seen in similar systems
[22]. A complex of Shc,
GRB2 and SOS may stimulate the small
G-protein Ras [23], leading
to activation of the
serine/threonine kinase Raf-1 [24-26]. Raf-1
is one of the activators
of the threonine/tyrosine kinase MEK1
[24, 25, 27] which
activates the serine/threonine MAP kinases
ERK1/2 [28]. In vsmc,
AngII activates ERK1/2 from about 2 to 30 min
[23, 25, 29-33].
Activation of ERK1/2 is terminated by the MKP-1 phosphatase
[34,
35].
and PLD, Src is probably responsible for
phosphorylation of
Shc in vsmc [18] as seen in similar systems
[22]. A complex of Shc,
GRB2 and SOS may stimulate the small
G-protein Ras [23], leading
to activation of the
serine/threonine kinase Raf-1 [24-26]. Raf-1
is one of the activators
of the threonine/tyrosine kinase MEK1
[24, 25, 27] which
activates the serine/threonine MAP kinases
ERK1/2 [28]. In vsmc,
AngII activates ERK1/2 from about 2 to 30 min
[23, 25, 29-33].
Activation of ERK1/2 is terminated by the MKP-1 phosphatase
[34,
35].
ERK1/2 can phosphorylate a number of proteins, at
least in vitro,
such as cytosolic phospholipase A2,
myelin basic protein,
transcription factors such as c-jun and
p62TCF and may increase
protein synthesis by activation of p90
ribosomal S6 kinase and
the mRNA cap-binding protein eIF-4A [1, 2, 36,
37]. Tyrosine
kinase inhibitors such as genistein or herbimycin
A reduced AngII-induced
activation of the ERK1/2 and JNK MAP
kinases, vessel contraction and
protein synthesis [1, 20,
38].
AngII-induced ERK1/2 activity in vsmc is dependent
on calcium
signalling since it was blocked by inhibition of PLC,
calmodulin
or intracellular calcium mobilization [23].
AngII-induced ERK1/2
activity was not affected by PKC inhibition
or down-regulation
with phorbol esters [23, 24]. However, antisense
inhibition of
the atypical PKC zeta subtype, which is not
down-regulated by phorbol
esters, decreased AngII-induced ERK1/2
activity [39].
AngII may also activate the signalling
pathways of other typical
tyrosine kinase-coupled receptors.
Thus, AngII induced transactivation
of the EGF receptor, without
EGF secretion [40], and stimulated
secretion of autocrine mediators
such as IGF-I [41]. These pathways
probably contribute to the
sustained activation of tyrosine kinases
by
AngII.
Reactive
oxygen species, such as superoxide
and hydrogen peroxide
(H2O2), can chemically
alter many cellular
components. However, oxidants once only known
for their toxicity
are now being recognized as possible signalling
molecules at
moderate concentration [42]. Inside the cell the
superoxide free
radical can be quickly metabolized by superoxide
dismutase (SOD)
to hydrogen peroxide which is then more slowly converted
to
water by catalase.
Oxidants may have both rapid and
long-term signalling effects.
Exogenous superoxide generated for
example with xanthine/xanthine
oxidase doubled IP3-induced
calcium release from the vsmc sarcoplasm.
This effect appeared
to be mediated by superoxide since it was
sensitive to SOD, but
not to catalase [42, 43]. H2O2
was also
capable of inducing rapid and sustained mobilization
of calcium
from agonist-sensitive stores in another vsm model
[44].
Exogenous superoxide increased DNA synthesis [45-48] and
cell
number [45, 48] in quiescent vsmc. Similarly, incubation with
H2O2
increased DNA synthesis [46-50] in the same
model. In some instances
the effect of exogenous oxidants may be
indirect, since it may
be dependent upon IGF-I, bFGF or the EGF
receptor [47, 51, 52].
The physiological relevance of treatments
with
exogenous oxidants was recently established by the discovery
of
an AngII-activated endogenous superoxide-generating pathway
in
vsmc [53]. The source of superoxide is a membrane-bound NADH
or
NADPH-dependent oxidase with features similar to the
phagocyte
NADPH oxidase [53-58]. This latter enzyme complex is composed
of a
catalytic membrane-bound cytochrome b558
heterodimer (gp91- and
p22-phox) that is activated by binding at
least three cytosolic components
(p47-, p67-phox and Rac2).
p47phox may be activated by protein
kinase C [59, 60] and Rac2
by exchange of GTP for GDP (Fig. 2).
AngII induced almost a
3-fold increase in superoxide production
and a 5-fold accumulation of
H2O2 in vsmc [57, 61].
Superoxide was the major
source of H2O2 because inhibition
of oxidase activity
with diphenylene iodonium (DPI) or p22-phox
antisense mRNA
inhibited the formation of both compounds [33,
53, 57, 61, 62].
This pathway was critical to growth in vsmc
since inhibition of
the oxidase or SOD, or overexpression of catalase,
greatly
reduced AngII-induced protein synthesis [53, 57,
61].
The function of oxidants was confirmed in-vivo.
Hypertension
induced by infusion of AngII (but not
norepinephrine) in the
rat was accompanied by up-regulation of
p22phox mRNA, cytochrome
b558 protein, and NAD(P)H oxidase-dependent
activity, as well
as an impairment of endothelium-dependent
relaxation. Importantly,
AngII-induced hypertension was
corrected by infusion of heparin-binding
SOD. Moreover, the
biochemical effects of AngII infusion were
inhibited in vitro by
DPI or liposomal SOD [58, 63, 64].
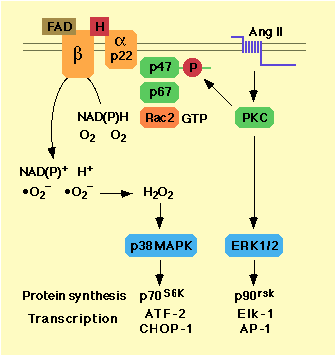
Figure 2. The NAD(P)H signalling cascade in vsmc
Exogenous superoxide increased the activity of ERK1/2, an effect that was inhibited by the superoxide scavenger tiron and protein kinase C down-regulation [48]. The effect of exogenous H2O2 on ERK1/2 activation has been controversial. ERK1, ERK2 or both were phosphorylated by exposure to H2O2 in some studies [51, 65, 66], but not in others [33, 48, 67]. Stimulation of vsmc with AngII increases endogenous H2O2 accumulation and is accompanied by ERK1/ERK2 activation [33, 67]. However, the fact that AngII-induced ERK1/2 phosphorylation was not inhibited by DPI, tiron or by overexpression of catalase suggests that these kinases are not redox-sensitive within cells [33, 48]. Exogenous H2O2 increased phosphorylation and/or activity of other MAP kinases present in vsmc, namely JNK, p38MAPK and BMK1 (ERK5) [33, 66, 67]. AngII induced p38MAPK activation appeared to result from endogenous H2O2 generation via the NAD(P)H oxidase pathway, since it was blocked by DPI or catalase overexpression [33]. It is worth noting that ERK1/2 and p38MAPK control together the major portion of AngII-mediated hypertrophy. While specific inhibitors of either kinase were partially effective, simultaneous incubation with both compounds blocked most of AngII-induced protein synthesis [33]. This may be explained by the fact that these two kinases regulate complementary sets of proteins and transcription factors. For example, p70 and p90 ribosomal S6 kinases are phosphorylated by p38MAPK and ERK1/2, respectively. Similarly, ATF-2 and CHOP-1 are activated by p38MAPK, while Elk-1 and AP-1 are stimulated by ERK1/2 (Fig. 2).
Following an immediate stimulation of vascular smooth muscle contraction, AngII chronically induces the cellular hypertrophy and proliferation observed in vascular diseases. These effects are mediated by a vast array of signals, notably heterotrimeric and small G-proteins, phospholipases, and kinase cascades which control gene transcription. It is interesting to note that the most recently recognized of these mediators, superoxide and H2O2, now fulfill the conditions required for classification as signalling molecules. Both agents are rapidly synthesized in response to agonist stimulation, have specific molecular targets and are rapidly degraded enzymatically. Although formerly known for their toxicity, these oxidants are now gaining acceptance as redox signal mediators. Their complex roles in AngII responsiveness are only beginning to be understood.
1. Berk,
B. C. and Corson, M. A. (1997) Circ Res
80: 607-16
2.
Griendling, K. K., Ushio-Fukai, M., Lassègue, B. et al.
(1997)
Hypertension 29: 366-73
3. Kai, H., Fukui, T., Lassegue,
B. et al. (1996) Mol Pharmacol 49:
96-104
4. Rhee, S. G. and
Bae, Y. S. (1997) J Biol Chem 272: 15045-8
5. Marrero, M. B.,
Paxton, W. G., Duff, J. L. et al. (1994) J Biol Chem
269:
10935-9
6. Marrero, M. B., Schieffer, B., Paxton, W. G. et al. (1995) J
Biol Chem
270: 15734-8
7. Touyz, R. M. and Schiffrin, E. L.
(1997) Hypertension 30: 222-9
8. Di Salvo, J. and Nelson, S. R.
(1998) FEBS Lett 422: 85-8
9. Venema, R. C., Ju, H., Venema, V.
J. et al. (1998) J Biol Chem 273:
7703-8
10. Schelling, J. R.,
Nkemere, N., Konieczkowski, M. et al. (1997) Am J
Physiol 272:
C1558-C66
11. Ushio-Fukai, M., Griendling, K. K., Akers, M. et al.
(1998) J Biol Chem
273: 19772-7
12. Lassègue, B.,
Alexander, R. W., Clark, M. et al. (1991) Biochem.
J. 276:
19-25
13. Lassègue, B., Alexander, R. W., Clark, M. et al.
(1993) Biochem.
J. 292: 509-17
14. Ushio-Fukai, M., Alexander,
R. W., Akers, M. et al. (in press) Mol Pharmacol
15. Exton, J. H.
(1997) J Biol Chem 272: 15579-82
16. Ishizaka, N., Griendling,
K. K., Lassegue, B. et al. (1998) Hypertension
32: 459-66
17.
Kosaka, T. and Gilman, A. G. (1996) J Biol Chem 271: 12562-7
18.
Linseman, D. A., Benjamin, C. W. and Jones, D. A. (1995) J Biol
Chem
270: 12563-8
19. Ishida, M., Marrero, M. B., Schieffer, B.
et al. (1995) Circ Res 77:
1053-9
20. Ishida, M., Ishida, T.,
Thomas, S. M. et al. (1998) Circ Res 82:
7-12
21. Schieffer, B.,
Paxton, W. G., Chai, Q. et al. (1996) J Biol Chem
271:
10329-33
22. Wan, Y., Kurosaki, T. and Huang, X. Y. (1996)
Nature 380: 541-4
23. Eguchi, S., Matsumoto, T., Motley, E. D.
et al. (1996) J Biol Chem 271:
14169-75
24. Liao, D. F., Duff,
J. L., Daum, G. et al. (1996) Circ Res 79:
1007-14
25. Molloy,
C. J., Taylor, D. S. and Weber, H. (1993) J Biol Chem
268:
7338-45
26. Schieffer, B., Drexler, H., Ling, B. N. et al.
(1997) Am J Physiol 272:
C2019-30
27. Ishida, Y., Kawahara, Y.,
Tsuda, T. et al. (1992) FEBS Lett 310:
41-5
28. Marrero, M. B.,
Schieffer, B., Li, B. et al. (1997) J Biol Chem
272:
24684-90
29. Malarkey, K., McLees, A., Paul, A. et al.
(1996) Cell Signal 8:
123-9
30. Tsuda, T., Kawahara, Y., Shii,
K. et al. (1991) FEBS Lett. 285:
44-8
31. Tsuda, T., Kawahara,
Y., Ishida, Y. et al. (1992) Circ Res 71:
620-30
32. Duff, J.
L., Berk, B. C. and Corson, M. A. (1992) Biochem Biophys Res
Commun
188: 257-64
33. Ushio-Fukai, M., Alexander, R. W., Akers, M. et
al. (1998) J Biol Chem
273: 15022-9
34. Duff, J. L., Marrero, M.
B., Paxton, W. G. et al. (1993) J Biol Chem
268: 26037-40
35.
Duff, J. L., Monia, B. P. and Berk, B. C. (1995) J Biol Chem
270:
7161-6
36. Duff, J. L., Marrero, M. B., Paxton, W. G. et
al. (1995) Cardiovasc
Res 30: 511-7
37. Giasson, E. and Meloche,
S. (1995) J Biol Chem 270: 5225-31
38. Leduc, I., Haddad, P.,
Giasson, E. et al. (1995) Mol Pharmacol 48:
582-92
39. Liao, D.
F., Monia, B., Dean, N. et al. (1997) J Biol Chem
272:
6146-50
40. Eguchi, S., Numaguchi, K., Iwasaki, H. et al.
(1998) J Biol Chem 273:
8890-6
41. Delafontaine, P. and Lou, H.
(1993) J Biol Chem 268: 16866-70
42. Suzuki, Y. J., Forman, H.
J. and Sevanian, A. (1997) Free Radic Biol
Med 22: 269-85
43.
Suzuki, Y. J. and Ford, G. D. (1992) Am J Physiol 262:
H114-6
44. Roveri, A., Coassin, M., Maiorino, M. et al. (1992) Arch
Biochem Biophys
297: 265-70
45. Li, P. F., Dietz, R. and von
Harsdorf, R. (1997) Circulation 96:
3602-9
46. Rao, G. N. and
Berk, B. C. (1992) Circ Res 70: 593-9
47. Delafontaine, P. and
Ku, L. (1997) Cardiovasc Res 33: 216-22
48. Baas, A. S. and
Berk, B. C. (1995) Circ Res 77: 29-36
49. Stauble, B.,
Boscoboinik, D., Tasinato, A. et al. (1994) Eur J Biochem
226:
393-402
50. Fiorani, M., Cantoni, O., Tasinato, A. et al. (1995)
Biochim Biophys
Acta 1269: 98-104
51. Rao, G. N. (1996) Oncogene
13: 713-9
52. Herbert, J. M., Bono, F. and Savi, P. (1996) FEBS
Lett 395: 43-7
53. Griendling, K. K., Minieri, C. A.,
Ollerenshaw, J. D. et al. (1994)
Circ. Res. 74: 1141-8
54.
Mohazzab, K. M. and Wolin, M. S. (1994) Am J Physiol 267:
L823-31
55. Mohazzab, K. M. and Wolin, M. S. (1994) Am J Physiol
267: L815-22
56. Pagano, P. J., Ito, Y., Tornheim, K. et al.
(1995) Am J Physiol 268:
H2274-80
57. Ushio-Fukai, M., Zafari,
A. M., Fukui, T. et al. (1996) J Biol Chem
271: 23317-21
58.
Rajagopalan, S., Kurz, S., Munzel, T. et al. (1996) J Clin Invest
97:
1916-23
59. Rotrosen, D., Yeung, C. L., Leto, T. L. et al.
(1992) Science 256:
1459-62
60. Jones, O. T. (1994) Bioessays
16: 919-23
61. Zafari, A. M., Ushio-Fukai, M., Akers, M. et al.
(1998) Hypertension
32: 488-95
62. Fukui, T., Lassegue, B., Kai,
H. et al. (1995) Biochim Biophys Acta
1231: 215-9
63. Fukui, T.,
Ishizaka, N., Rajagopalan, S. et al. (1997) Circ Res
80:
45-51
64. Bech Laursen, J., Rajagopalan, S., Galis, Z. et
al. (1997) Circulation
95: 588-93
65. Guyton, K. Z., Liu, Y.,
Gorospe, M. et al. (1996) J Biol Chem 271:
4138-42
66. Zhang,
J., Jin, N., Liu, Y. et al. (1998) Am J Respir Cell Mol Biol
19:
324-32
67. Abe, J., Kusuhara, M., Ulevitch, R. J. et al. (1996) J Biol
Chem 271:
16586-90